
高效光催化材料的设计与制备
2017-02-14 03:07王泽岩黄柏标
中国材料进展 2017年1期
王泽岩,黄柏标,戴 瑛
(1.山东大学 晶体材料研究所,山东 济南 250100) (2.山东大学物理学院,山东 济南 250100)
特约专栏
高效光催化材料的设计与制备
王泽岩1,黄柏标1,戴 瑛2
(1.山东大学晶体材料研究所,山东 济南 250100) (2.山东大学物理学院,山东 济南 250100)
光催化技术是解决能源短缺和环境污染问题的最有效解决途径之一,近期引起了世界各国科学家们的广泛关注。然而,目前光催化材料较低的转化效率,严重制约了光催化技术的进一步发展和实际应用。因此,设计和制备新型高效光催化材料具有重要意义。我们基于材料晶体结构设计,并结合材料微结构调控和制备技术,从拓展光催化材料的光吸收范围和提高光生载流子分离效率两个制约光催化效率的最主要问题入手,提出了一些新型高效光催化材料的设计理论和制备方法。主要围绕表面等离子体光催化材料、红外光催化材料、极性光催化材料等几种新型光催化材料,对材料设计理论、制备方法、结构与性能之间的关系及光催化微观机理几个方面进行了详细讨论。
光催化;光吸收;载流子分离;材料设计与制备
1 光催化技术的发展现状
近年来,随着我国经济的飞速发展和工业化水平的不断提高,对能源的消耗也日益增大。虽然我国的化石能源储量相对丰富,但依然无法满足日益增长的能源需求。目前,除煤炭外,我国其他化石能源,如:石油、天然气等主要依赖大量进口,能源短缺问题日趋严峻。与此同时,化石能源的大量消耗引起的环境污染问题同样引起越来越多人的重视。我国以煤炭为主的能源结构和日益增长的工业化生产规模,使得人们赖以生存的自然环境遭到严重破坏。目前,我国已经成为世界上污染最严重的国家之一。能源短缺和环境污染问题已经成为制约我国未来经济发展和社会稳定的关键问题之一。因此,如何有效解决我国严峻的能源短缺和环境污染问题迫在眉睫。
光催化技术是一种能够有效利用太阳能,并将低密度的太阳能转化为高密度的化学能的新技术。基于光催化技术,可利用太阳能实现分解水产生氢气和氧气、将二氧化碳还原为有机燃料、降解水体和空气中的有机污染物等。相对于目前解决能源与环境问题的其他解决方案,光催化技术不仅能够将太阳能转化为氢能、有机燃料等新能源,还能将环境中的有机污染物转换为对环境友好的二氧化碳和水,不产生二次污染,从而解决环境污染问题。因此,光催化技术被认为是一种能够有效解决人类社会未来能源短缺和环境污染问题的最有效手段之一,引起了世界各国科学家们的广泛关注,成为能源与环境研究领域的热点之一。
自20世纪70年代,日本科学家利用二氧化钛和紫外光实现光催化分解水产生氢气和氧气以来,光催化技术进入了一个飞速发展的阶段。在过去的几十年时间里,世界各国科学家在光催化领域进行了大量的研究。我国开始进行光催化技术研究起步较早,目前我国在光催化技术领域的研究水平,整体处于较高水平。目前,光催化材料种类大大丰富,光催化效率也得到了大幅提高。然而,目前光催化效率依然无法满足实际生产生活的要求,相对较低的光催化转化效率成为制约光催化技术广泛应用的最主要问题。因此,进一步提高光催化材料的催化效率依然是光催化技术领域所面临的最主要问题。
2 高效光催化材料的设计思路
从光催化反应过程来看,光催化可分为两步,如图1所示。首先,当太阳光照射到半导体材料表面时,大于半导体禁带宽度的光子能量被半导体材料吸收。吸收的光子能量将半导体价带中的电子激发到导带,同时在价带中产生相同数量的空穴,产生光生电子-空穴对。然后,光生电子与空穴对,有一部分在半导体内部或表面迅速复合,以热量或发光的形式将能量消耗,而未复合的光生电子-空穴对扩散到半导体材料表面参与氧化还原反应。由此可见,在光催化过程中,影响光催化转化效率的最主要因素主要是:光催化材料的光吸收范围和光生载流子的分离与利用效率。因此,进一步拓展光催化材料的光谱吸收范围和提高光生载流子分离与利用效率是进一步提高光催化转化效率的关键。
为解决这两个关键问题,世界各国科学家们经过长期大量研究已经探索了一系列的解决方案。例如:为解决光催化材料光吸收范围较窄的问题,人们开发了通过对半导体材料进行金属元素或非金属元素掺杂、形成固溶体等手段,通过对半导体材料的能带结构进行调控,从而拓展光催化材料的光吸收范围;或者利用有机染料、量子点等敏化剂来拓展光催化材料的光吸收范围。为促进光生载流子的有效分离,通过将能带结构匹配的半导体材料复合形成II型异质结,或者通过负载贵金属纳米颗粒等方式,利用不同材料形成的界面电场来促进光生载流子的有效分离与利用。虽然这些方法能够进一步拓展光催化材料的光吸收范围和提高光生载流子分离效率,从而提高光催化转化效率。然而,这些常规方法在提高光催化转化效率的同时通常存在一定的负面效果。如:通过元素掺杂能够缩短半导体能带宽度,但是杂质元素的引入,会在半导体内部引入缺陷,成为光生载流子的复合中心,从而降低光生载流子的分离效率;异质结的界面电场虽然能够促进光生载流子的分离,然而界面处容易形成缺陷,并且界面电场作用范围较窄、强度较小,并不能有效提高光生载流子的分离效率等。因此,如何进一步拓展光催化材料的光谱吸收范围,同时进一步促进光生载流子的分离,是设计和制备新型高效光催化材料的关键。
由材料的结构与性质之间的关系可知,材料的物理性质同材料的晶体结构之间存在直接联系。从晶体结构出发,结合晶体结构设计,通过调节和控制材料晶体结构,从而调控材料电子结构,最终调节材料的光吸收、载流子传输等物理性能,是能够有效改善光催化材料光吸收范围和光生载流子分离效率的有效手段。因此,本文从晶体结构设计入手,通过晶体结构和电子结构调控,设计和探索具有宽光谱吸收和高载流子分离效率的新型高效光催化材料。
3 高效光催化材料的设计与制备
由于光催化材料的太阳能转换效率主要由光催化材料的光吸收范围和光生载流子的分离效率所决定。因此,进一步拓展光催化材料光谱吸收范围和提高光生载流子的分离与利用效率是进一步提高光催化材料太阳能转化效率的关键。不同于现有用于提高光催化材料太阳能转化效率的常规手段,作者课题组结合在晶体结构与电子结构设计、材料制备与微结构调控、理论计算与模拟等方面的研究基础和特色,提出了一系列具有创新性的新型高效光催化材料的设计理论,并制备了系列具有宽光谱响应范围和高载流子分离效率的新型高效光催化材料,如:表面等离子体光催化材料、红外光催化材料、极性光催化材料等。
3.1 表面等离子体光催化材料
表面等离子体共振效应是贵金属纳米颗粒(如:Au, Ag)的独特性质之一,在分子成像、表面增强拉曼光谱、太阳能电池等众多领域具有广泛的应用。当入射光频率同贵金属导带电子的共振频率相同时,贵金属纳米颗粒导带电子同入射光子产生谐振,从而对该频率的光表现出强烈的光吸收。通过调节贵金属纳米颗粒的尺寸、形貌及其周围介质环境可连续调节其光吸收范围。因此,相对于半导体材料,贵金属纳米颗粒的表面等离子体效应对光的吸收,范围更宽、强度更大。可以有效解决光催化材料光吸收范围窄的问题。
2008年,作者团队首次将卤化银材料应用于光催化领域,利用离子交换和光还原法制备了Ag@AgCl表面等离子体光催化材料[1]。由于Ag纳米颗粒的表面等离子体共振效应,Ag@AgCl表面等离子体光催化材料在整个可见光区域表现出强烈的光吸收,并在可见光照射下降解有机染料过程中表现出极其优异的催化性能(如图2所示)。基于贵金属纳米颗粒的表面等离子体共振效应和金属-半导体接触,提出了新型卤化银基表面等离子体光催化材料的概念,从而开辟了一条拓展光催化材料可见光吸收,促进光生载流子分离,提高光催化活性的新途径。
以此为基础,作者课题组围绕Ag@AgX(X=Cl, Br, I)表面等离子体光催化材料,结合材料复合、微结构调控、材料制备手段,进行了一系列研究。制备了包括:不同形貌的Ag@AgX(X=Cl, Br)、Ag@Ag(Br,I)、Ag@Ag(Cl,Br)、Ag@AgCl-AgI、Ag/AgBr/WO3·H2O、Ag/AgBr/BiOBr、AgI/BiOI等系列具有宽可见光吸收的高效表面等离子体光催化材料[2-14]。此外,作者课题组在Ag@AgX表面等离子体光催化材料的研究基础上,进一步研究了M@TiO2(M=Ag, Au, Pt)表面等离子体光催化材料及其选择性有机光合成方面的性质[15]。由于表面等离子体光催化材料在拓展光催化材料光吸收和提高光催化活性方面的巨大优势,使其成为近期光催化领域的研究热点之一,受到了国内外同行的广泛关注。
为进一步探索表面等离子体光催化材料的微观机制,作者团队结合理论计算模拟对Ag@AgX基表面等离子体光催化材料和M@TiO2表面等离子体光催化材料中,贵金属纳米颗粒尺寸形貌等对表面等离子体共振效应的影响机制、表面等离子体效应的能量传输机制、表面局域电磁场及载流子传输过程与机理进行了详细研究[16-18]。并在近期受NPG子刊Light:Science&Applications邀请为其撰写了相关综述[19]。
虽然,表面等离子体光催化材料能够极大拓展光催化材料的光吸收范围,从而有效提高光催化效率。然而,基于贵金属纳米颗粒的表面等离子体光催化材料,受限于贵金属高昂的价格,在实际应用中存在较大限制。近期,作者团队的理论计算结果表明,氧化物半导体材料通过高浓度的H掺杂,能够大大提高半导体材料的载流子浓度,从而表现出与贵金属纳米颗粒类似的表面等离子体共振效应[20]。这为探索非贵金属表面等离子体光催化体系提供了一个新的思路。然而,由于氧化物半导体材料中产生表面等离子体效应所需的载流子浓度较高,普通的H掺杂手段,很难满足这一要求。因此,寻找能够产生高H掺杂浓度的实验制备方法是实现非贵金属表面等离子体光催化材料的关键。
近期,作者课题组利用“H-Spillover Process”,通过将Pd负载到MoO3表面,然后进行H化处理,在室温下成功制备了高H掺杂的HxMoO3非金属表面等离子体光催化材料[21]。由于Pd对H2良好的催化解离性质,能够将H2转化为活性H原子,从而进入MoO3材料内部,产生高H掺杂的HxMoO3。如图3所示,HxMoO3在565 nm表现出强烈的光吸收。相比之下,纯的MoO3在300 ℃条件下氢化,其吸收光谱几乎没有任何变化。结合XPS对氢化前后样品的结构进行分析发现,氢化后,Mo6+的价态被部分还原至Mo5+和Mo4+,Pd2+被还原至Pd0。

图3 氢化前后MoO3样品的吸收光谱(a),Mo 3d的XPS峰(b),Mo的K边XANES和(c)FT-EXAFS(d)Fig.3 UV-Vis diffuse reflectance spectra (a), Mo 3d XPS spectra (b), Mo K-edge XANES (c)and FT-EXAFS spectra (d) for Pd/MoO3 products before and after H2 reduction at room temperature
通过对HxMoO3样品在不同温度下氧化,可以发现其吸收峰位置及宽度发生相应的变化。当在100 ℃和200 ℃退火后,HxMoO3的吸收峰由565 nm红移至588 nm,且吸收峰展宽。XRD分析显示样品的结构由开始的H1.68MoO3变为H0.9MoO3。这表明,随着在空气中氧化温度的提高,样品中H掺杂浓度减少,从而使样品中载流子浓度降低,使表面等离子体共振频率随之降低,造成吸收光谱的红移。由此可见,可通过氧化程度的不同,调节HxMoO3体系中H的掺杂浓度,从而实现对其表面等离子体共振吸收的可控调节。
同MoO3相似,WO3在Pd的辅助下,同样能够在室温下实现高浓度的H掺杂,在可见光区域产生表面等离子体共振吸收。然而,WO3的表面等离子体共振吸收位置位于660 nm。由于表面等离子体共振频率同材料的载流子浓度直接相关,因此,HxMoO3和HxWO3的表面等离子体共振吸收峰位置的不同,表明材料中载流子浓度之间的差异。
为进一步探索MoO3和WO3中H离子掺杂浓度不同的围观机制,作者课题组对其进行了理论计算模拟。计算结果显示,MoO3的层状结构更有利于H离子的进入,H离子进入MoO3的层间位置,并同O原子键合,同时将本身的电子通过O原子传递给Mo原子。由于H离子贡献的电子处于完全离域化状态,因此能够为体系提供较高的自由电子。而WO3材料为变形的ReO3型晶体结构,WO6八面体以共定点的方式连接。因此,H原子进入WO3中孔隙位置。但是,由于H原子提供的电子部分局域化,因此限制了对体系提供自由电子的数量。相对于MoO3、WO3中的载流子浓度较低。由此可见,H原子在氧化物半导体中的位置和键合状态对体系的载流子浓度具有重要影响。为验证这一理论,作者团队选择V2O5进行了对比试验。由于V2O5中H原子以间隙H状态存在,因此,H原子完全处于局域化状态,不能为体系提供自由电子,从而使其不能实现表面等离子体共振效应。
这一工作,为制备非贵金属表面等离子体光催化材料提供了一种新的方法,并为设计新型非贵金属表面等离子体光催化材料提供了相关的设计理论。
3.2 红外光催化材料
为进一步提高光催化材料的催化活性,人们在拓展光催化材料的光吸收范围方面进行了大量的研究。虽然,目前光催化材料的光吸收范围已从紫外光区域拓展至可见光。然而,占太阳光谱近一半能量的红外光却没有得到充分的利用。由于半导体材料对光子能量的吸收主要由半导体能带跃迁引起。因此,能够吸收红外光能量的半导体材料需要极窄的能带宽度。这样同时降低了半导体材料的氧化-还原能力。为解决这一问题,近期人们将具有可见光响应的半导体材料同上转换材料复合,通过上转换材料将红外光转化为可见光,驱动可见光光催化材料进行光催化反应。然而,受限于较低的上转换能量转换效率(约1%),这种基于上转换材料的红外光催化材料并不能有效利用红外光能量。因此,探索具有红外光响应的新型高效光催化材料具有重要的意义。
近期,作者团队结合晶体结构设计理论,设计并制备了Cu2(OH)PO4红外光催化材料[22]。如图4a所示,Cu2(OH)PO4在可见光和近红外区域具有强烈的光吸收。通过Gaussian拟合,可将吸收光谱拟合成位于670, 864, 1121和1349 nm处的4个吸收峰。为研究Cu2(OH)PO4的光催化活性,我们在红外光(λ>800 nm)照射下进行了2,4-DCP水溶液的降解实验。如图4b所示,Cu2(OH)PO4在6个小时里,能够降解90%的2,4-DCP。通过XPS分析对光催化反应前后Cu2(OH)PO4中Cu的价态进行分析,发现Cu的价态没有任何变化,证明了该材料的稳定性。为探索和研究Cu2(OH)PO4具有红外光催化活性的基本原理,作者所在课题组对该材料的晶体结构和电子结构进行了详细的研究。Cu2(OH)PO4的晶体结构如图5所示,

图4 (a) Cu2(OH)PO4的紫外-可见-近红外吸收光谱,(b) 红外光照射下(λ> 800 nm)降解2,4-DCP的自挥发曲线(1)和降解曲线(2)Fig.4 UV-Vis-NIR absorption spectra of Cu2(OH)PO4 (a) and comparison plots of the self-volatilization(1) and photodegradation (2) of 2,4-DCP in aqueous solution over Cu2(OH)PO4 with NIR irradiation (λ> 800 nm) (b)
在Cu2(OH)PO4材料中存在两种不同配位的Cu离子,这两种Cu离子分别同周围的O原子结合形成CuO4(OH)三角双锥和CuO4(OH)2八面体结构,并且它们之间以氧桥的方式连接。通过理论计算结果发现,在Cu2(OH)PO4中,其价带和导带均由O 2p和Cu 3d轨道组成。而导带由4个分离的非占据态组成,价带同导带的4个非占据态之间的电子跃迁使其对近红外光具有强烈的吸收,对应于图4中吸收光谱拟合出的4个红外吸收峰。当电子从价带激发至导带时,首先被激发至导带位置最高的非占据态,然后逐渐衰减至位置较低的非占据态。而从Cu2(OH)PO4的态密度图中可以看出,位置较高的非占据态主要由CuO4(OH)三角双锥的Cu 3d和O 2p构成,而位置较低的非占据态主要由CuO4(OH)2八面体结构的Cu 3d和O 2p构成。这表明光生电子会从CuO4(OH)三角双锥向CuO4(OH)2八面体转移。由于CuO4(OH)三角双锥同CuO4(OH)2八面体之间通过共顶点的方式通过OH连接,通过计算OH中O 2p轨道对4个非占据态的贡献发现,OH对位置较低的非占据态贡献较小,而对位置较高的非占据态贡献较大。因此,当光生电子从CuO4(OH)三角双锥传递到CuO4(OH)2八面体之后将不会反向传输,从而实现了光生载流子的有效分离。
在光催化过程中,如方程(1)~(4)所示,Cu2(OH)PO4吸收光子能量之后,光生电子从CuO4(OH)三角双锥传递到CuO4(OH)2八面体,CuO4(OH)三角双锥和CuO4(OH)2八面体中的CuII离子在失去和得到电子之后,分别被氧化和还原为CuIII和CuI。CuIII具有较强的氧化能力可将有机物降解,同时CuIII再次转变成CuII。而CuI同H+和O2作用也重新转变为CuII,完成光催化反应过程。
TBP CuII+ hv = TBP CuIII+ e-
(1)
OCT CuII+ e-= OCT CuI
(2)
TBP CuIII+ 2,4-DCP = TBP CuII+ H2O + CO2+
H++ Cl-
(3)
OCT CuI+ H++ O2= OCT CuII+ H2O
(4)

图5 Cu2(OH)PO4的晶体结构(a),态密度分布图(b),OH基团的O 2p、八面体和三角双锥中Cu 3d轨道的能带分布图(c),八面体和三角双锥中O原子的O 2p轨道态密度分布图(d)Fig.5 The crystal structure of Cu2(OH)PO4(a), the PDOS plots for the Cu 3d and O 2p states (b), the PDOS plots for the O 2p states of the OH groups,the Cu 3d states of the octahedral Cu sites and those of the trigonal bipyramidal Cu sites (c), the PDOS plots for the O 2p s tates of the O atoms belonging only to the CuO4(OH)2 octahedra and those belonging only to the CuO4(OH) trigonal bipyramids (d)
这种基于不同配位的金属离子间的电荷转移过程,不仅能够有效拓展光催化材料的近红外光吸收,同时能够有效的促进光生载流子的分离,有效提高光催化活性。因此,为探索和设计新型高效的红外光响应光催化材料提供了一条新的设计思路。该工作发表后,美国化学学会C&E News对作者课题组所做的工作进行了高度评价,称其是至今为止第一个真正的具有近红外光活性的光催化材料,对探索新型太阳光驱动的光催化材料开辟了一条新的途径。随后,作者团队结合理论计算,对Cu2(OH)PO4材料进行了详细的理论研究,进一步揭示了其光吸收和电荷转移机理[23]。
在Cu2(OH)PO4基础上,作者课题组探索了系列具有相似结构的材料,如:Cu3(OH)4SO4、Cu4(OH)6SO4、Cu2(OH)3Cl[24]。同Cu2(OH)PO4类似,Cu3(OH)4SO4、Cu4(OH)6SO4、Cu2(OH)3Cl中均具有两种不同配位的Cu离子,并且在近红外光区域均具有强烈的光吸收能力。然而,在光催化过程中发现,Cu3(OH)4SO4和Cu4(OH)6SO4具有红外光催化活性,而Cu2(OH)3Cl没有。根据密度泛函理论详细的分析了Cu3(OH)4SO4、Cu4(OH)6SO4、Cu2(OH)PO4和Cu2(OH)3Cl的电子结构。研究发现,虽然Cu2(OH)3Cl中同其他材料类似,都具有Cu-O-Cu′的桥连结构,但是相对于SO4和PO4,Cl没有空能级与O原子结合,从而不能够容纳电子,从而使Cu2(OH)3Cl的基态和激发态中,两个Cu离子的电子分布没有任何变化,使其没有任何光催化活性。因此,基于该工作,作者课题组可以得到以下规律:材料中需由两种不同配位环境的Cu2+构成,形成具有CuOm和Cu′On不同多面体配位,并且由Cu-O-Cu′连接。同时,体系中需要存在电子受体基团(SO4或者PO4)与他们的CuOm和Cu′On相连,使其能够有效捕获光生电子,进而使光生电子和空穴发生分离,满足光催化条件。
关于红外光催化材料的研究,揭示了材料内部不同配位环境的金属离子间的电荷转移,不仅能够大大拓展光催化材料的光吸收范围至近红外区域,还能够有效促进光生载流子的有效分离,为探索具有红外光响应的新型光催化材料提供了新的设计理论和思路。
3.3 极性光催化材料
在光催化过程中,半导体材料吸收光子能量所产生的电子与空穴,从本质上讲是带有负电和正电的电荷。因此,电场是促使光生电子和空穴有效分离的最佳驱动力。目前,人们为促进光生载流子分离所普遍采用的方法是通过将具有合适能带结构的不同半导体材料复合形成II型异质结,或者将贵金属纳米颗粒负载到半导体光催化材料表面,通过不同材料的载流子浓度不同,形成界面电场,作为促进光生载流子分离的驱动力。但是,由于半导体异质结或贵金属负载产生的界面电场,仅存在于材料界面一个很小的范围内,且电场强度相对较小,因此对光生电子和空穴的分离效率有限。此外,由于不同材料之间的界面更容易形成缺陷,成为光生载流子的复合中心,因此通过构建异质结或贵金属负载的方式,并不能有效的提高光生载流子的分离效率。
然而,基于材料各向异性,在众多材料中,极性材料其晶体结构中正负电荷的中心并不重合,从而在材料内部产生自发极化。这种由于自发极化产生的内建电场存在于整个材料内部,相对于异质结或贵金属负载形成的界面电场,这种自发极化产生的内建电场作用范围更大,且电场强度更强。而单一材料内部更容易通过控制制备方法提高其结晶性,从而减少缺陷,降低光生载流子的复合几率。因此,相对于目前通过异质结和贵金属负载的方式来提高光生载流子分离效率的方法,极性材料的内建电场是一种能够促进光生载流子分离的更有效手段。因此,作者课题组近期在极性光催化材料方面进行了大量的研究。
作者课题组首先利用水热法制备了BiOIO3[25]。如图6所示,BiOIO3是由Bi2O22+层和IO3-层组成的层状结构,其中IO3-同Bi2O2层之间通过O原子项目连接。由于IO3-的定向排布,使其在沿c轴方向产生自发极化。当BiOIO3吸收光子能量产生光生电子和空穴时,在内建电场的作用下,电子和空穴将会向相反的方向移动,从而使其有效分离。为验证这一想法,作者课题组利用BiOIO3在紫外光照射下分解MO染料来表征其光催化活性。在紫外光照射下,BiOIO3能够在9 min内将MO染料完全降解,相比之下,P25在相同的时间内只能降解大约25%。为进一步验证BiOIO3的光催化活性提高主要由于内建电场的存在所引起的。作者课题组制备了同BiOIO3具有类似的层状结构,但不存在自发极化的材料,如:Bi2O2CO3、BiOBr、BiOI等。结果发现,虽然这些含Bi化合物都具有类似的结构,但其光催化活性远低于BiOIO3。为进一步排除IO3存在对光催化活性的影响,作者课题组还制备了含有IO3但不存在自发极化的AgIO3,结果表明IO3并不是引起BiOIO3光催化活性提高的主要原因。因此,通过实验验证,我们认为BiOIO3具有较高的光催化活性的原因主要可以归结于其IO3定性排列所引起的自发内建电场对光生载流子分离效率的提高。为进一步提供理论支持,我们结合理论计算,对BiOIO3的电子结构进行了详细的研究,并提出了相应的光催化机理。在BiOIO3研究的基础上,我们结合晶体结构设计理论探索了系列具有类似结构的Bi系极性光催化材料,如Bi2O2[BO2(OH)]、Bi4V2O11等[26-28]。

图6 BiOIO3的晶体结构示意图Fig.6 The perspective view of Bi2O2 and IO3 layer in BiOIO3
经过对极性光催化材料的相关研究,我们发现极性材料的极化内建电场能够有效促进光生载流子分离,进而提高光催化活性。然而,目前对于极性光催化材料的认识依然较浅,有很多基本问题依然不清楚,如:极性内建电场对于光生载流子分离效率有多大提高?内建电场方向对光生载流子的影响等。因此,进一步研究内建电场对光生载流子分离和传输机制和相关设计理论,是探索新型高效的极性光催化材料的关键。
由于极性材料的内建电场仅存在于材料内部,因此,为详细研究极性材料内建电场对光生载流子分离和传输机制的影响,我们选择利用ZnO单晶材料制备电极,通过光电化学的方法研究极性材料内部极化内建电场对光生载流子分离的影响[29]。ZnO是一种常见的半导体材料,其晶体结构为六方纤锌矿结构。由于ZnO晶体内部Zn和O原子沿c轴方向的周期性排布,使其在沿c轴方向存在一个由Zn-(0001)面指向O-(000-1)面的自发内建电场。因此,为研究内建电场对光生载流子分离效率的影响,我们通过控制单晶晶片内部内建电场方向,制备了3种不同暴露晶面的ZnO单晶光电极,如图7所示。当ZnO单晶电极的暴露面为Zn-(0001)面时(Zn-SC),内建电场方向垂直于电极表面指向内表面,当ZnO单晶电极暴露晶面为O-(000-1)面时(O-SC),内建电场方向垂直于电极表面指向外表面,而当ZnO单晶电极暴露面为M-(10-10)面时(M-SC),内建电场垂直于电极表面。同时,为了更好的比较单晶光电极同纳米光电极之间的区别,作者课题组还制备了ZnO纳米棒阵列光电极(ZnO NRs)。

图7 ZnO单晶光电极和ZnO纳米棒阵列电极示意图Fig.7 Schematic diagram of ZnO single-crystalline photoanodes and ZnO nanorods photoanodes
利用ZnO单晶光电极和ZnO纳米棒阵列电极进行光电化学分析可见,如图8所示。O-SC具有最高的光电流响应。当外加偏压为0.7 V(vsRHE)时,O-SC的光电流密度达到了1.47 mA/cm2。相比之下,M-SC、Zn-SC和ZnO NRs在相同偏压下的光电流密度仅为:0.7, 0.48和0.41 mA/cm2。与此同时,ZnO单晶光电极和ZnO纳米棒阵列光电极的能量转换效率和IPCE值表现出相似的规律。由于ZnO单晶光电极中,除内建电场方向不同之外,其他参数均相同。因此,造成ZnO单晶光电极性能差异的主要问题可归结于ZnO单晶电极内部内建电场方向不同。由于O-SC中,内建电场方向同光生载流子分离方向一致,因此内建电场的存在可以促进光生载流子的分离。反之,Zn-SC中,内建电场方向同光生载流子分离方向相反,因此,内建电场的存在使光生载流子向相反的方向移动,因此使得光生载流子的复合升高。而M-SC中,由于内建电场方向垂直于光生载流子的分离方向,因此,内建电场基本不对光生载流子的分离起到任何作用。所以,ZnO单晶光电极的光电化学活性存在O-SC>M-SC>Zn-SC的顺序。而ZnO NRs其暴露晶面主要为M-(10-10)晶面,其较高的缺陷浓度,使其光生载流子的复合几率远高于M-SC。因此,其光电流密度低于M-SC。
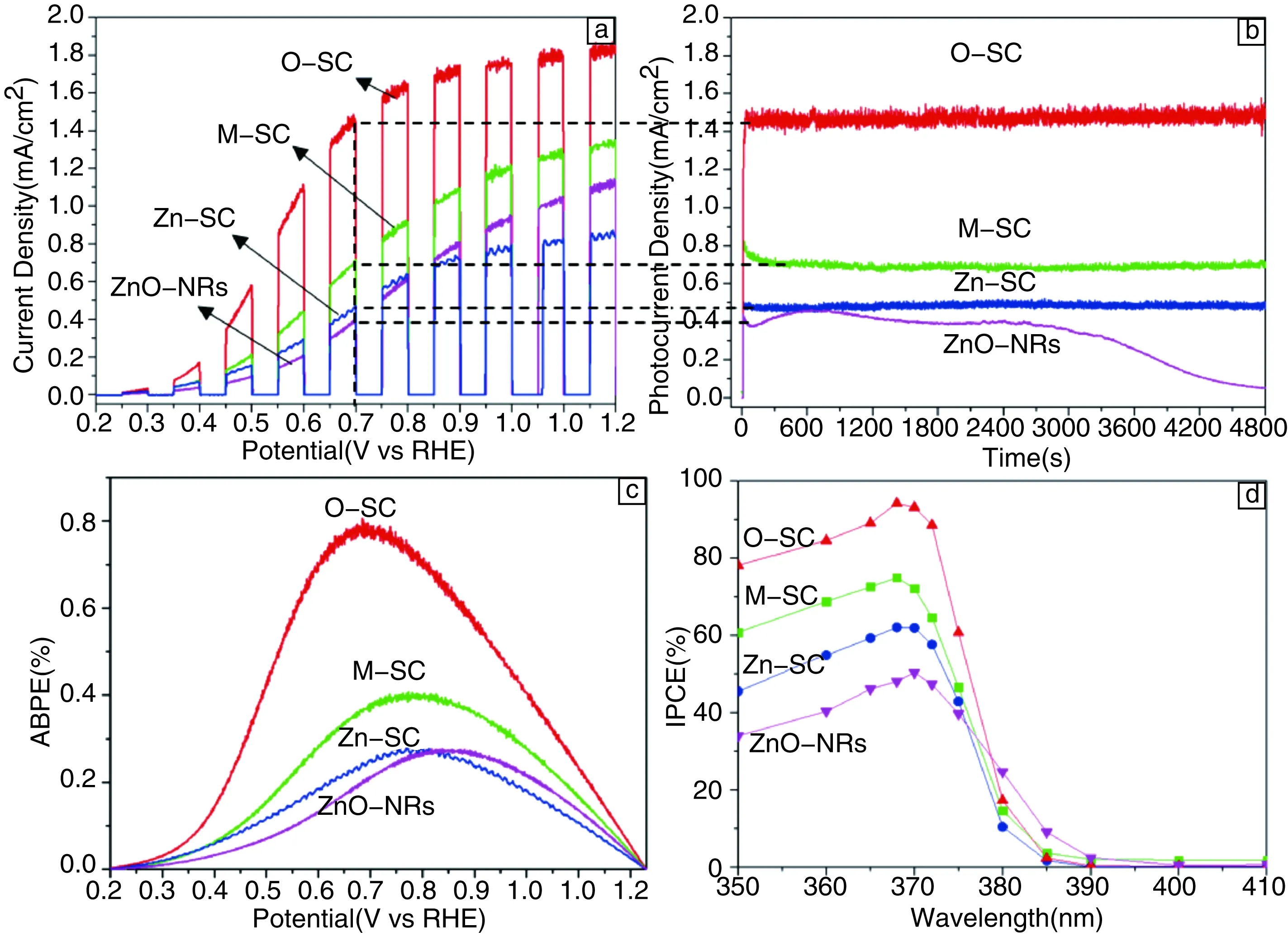
图8 ZnO单晶光电极和ZnO纳米棒阵列光电极的光电流曲线(a),光电流密度随时间变化曲线(b),施加偏压下的光电转化效率(c) 和不同波长的入射光的光电转换效率(d)Fig.8 I-V (a), I-t (b), ABPE (c) and IPCE (d) curves of ZnO single-crystalline photoanodes and ZnO nanorods photoanode
为进一步研究ZnO内部内建电场对光生载流子分离效率的影响,作者课题组分别研究了ZnO光电极内部载流子分离效率和表面载流子注入效率。研究结果表明,ZnO单晶光电极内部载流子分离效率同光电流密度曲线表现出类似的规律,O-SC>M-SC>Zn-SC。而相比之下, ZnO单晶光电极的表面载流子注入效率则差别不大。因此,我们推测,极性材料内部内建电场仅作用于材料内部,促进光生载流子在材料内部的分离。而由于极性材料的表面自稳定,内建电场在极性材料表面逐渐消失表现出电中性。因此,光生载流子在半导体/电解质界面分离率主要依赖于材料表面同电解质之间形成的界面电场。由此可见,极性材料的内建电场能够有效提高光生载流子分离效率,但仅限于极性材料内部,且依赖于内建电场的方向,当内建电场方向同载流子传输方向一致时促进其分离,反之则促进其复合。而内建电场对光生载流子的表面注入效率不起作用,光生载流子在界面的分离主要依赖于半导体材料同电解质界面处的界面电场。
基于对极性光催化材料的研究,我们发现极性光催化材料的内建电场能够有效促进光生载流子的分离,从而大大提高光催化活性。然而,由于材料内部自发极化电场的存在,使极性光催化材料的能带进一步拉宽,因此大大限制了极性光催化材料的光吸收范围。因此,如何拓展极性光催化材料的光吸收范围是设计和探索高效极性光催化材料的关键。
基于我们对红外光催化材料的研究,发现通过构建不同配位环境的金属离子,利用金属离子间的电荷转移,能够大大争抢材料对光子能量的吸收,拓展其光吸收范围。因此,结合红外光催化材料和极性光催化材料各自的优点,制备既具有宽光谱响应范围,又具有较高的载流子分离效率的新型极性光催化材料,能够进一步提高光催化活性。硅酸盐是世界范围内分布最广的矿物之一,其储量仅次于氧,且容易形成不同的配位环境,这为构建离子间电荷转移提供了一个良好的化学环境。而金属元素中,Ag由于其独特的d10原子结构,使其具有良好的可见光吸收。因此,我们结合硅酸盐和元素Ag各自特点,试图制备具有宽光谱响应范围和高载流子分离效率的硅酸银系列新型极性光催化材料。
首先,作者课题组利用简单的化学沉淀法制备了Ag6Si2O7[30]。如图9所示,该材料在整个可见光区域具有强烈的光吸收。在可见光照射下降解Mb染料过程中,Ag6Si2O7表现出极高的光催化活性,在10 min内可将染料完全降解,远高于具有高光催化活性的Ag3PO4和Ag2O。此外,Ag6Si2O7在波长大于700 nm的光照射下依然表现出较高的光催化活性。通过对Ag6Si2O7在光催化过程中所产生的活性物质进行表征,可以发现当加入Na2EDTA作为捕获剂时,其光催化活性消失,证明其光催化过程中的活性物质为光生空穴。

图9 Ag6Si2O7的吸收光谱(a),光催化降解Mb染料的降解曲线(b),不同滤波片的透过光谱(c),在不同波长的光照射下降解Mb染料的降解曲线(d)Fig.9 UV-Vis absorption spectra of Ag6Si2O7 (a), the degradation plots of Mb dye over Ag6Si2O7 under visible light irradiation (> 420 nm) (b), the transmission spectra of different filters (c), the degradation plots of Mb dye over Ag6Si2O7 under light irradiation with different filters (d)
通过对其晶体结构进行研究,发现在Ag6Si2O7中,SiO4四面体通过共顶点的方式连接形成Si2O7基团,而12个Ag+则形成3种不同配位的Ag+,并且不同配位的Ag之间通过Ag-O-Ag相互连接。通过对Ag6Si2O7内部化学键的偶极矩进行计算,发现在其内部存在一个沿b轴方向的内建电场。结合理论计算,对Ag6Si2O7的电子结构进行分析,发现由于不同配位的Ag离子对Ag6Si2O7导带贡献不同,使光生载流子能够在不同配位的Ag+之间进行传递,传递方向由四配位的Ag传递给三配位的Ag,最后传递给二配位的Ag。此外,电子在不同配位的Ag离子之间的传递方向恰好同Ag6Si2O7内部内建电场方向一致,从而使内建电场能够进一步促进光生电子和空穴的有效分离,从而进一步提高其光催化活性。因此,在Ag6Si2O7中,由于硅酸根的存在,为Ag离子提供了丰富的配位环境,从而导致其在整个可见光区域具有强烈的光吸收。与此同时,在内建电场和不同配位Ag离子之间的电荷转移共同作用下,其具有较高的光生载流子分离效率,从而使其在整个可见光区域具有较高的光催化活性。
由于硅酸根丰富的配位方式,基于Ag6Si2O7的研究结果,作者团队还研究了其他硅酸银系列极性光催化材料,如:非晶硅酸银、Ag9(SiO4)2NO3、Ag10Si4O13等新型硅酸银系列光催化材料[31-33]。这些硅酸银系列光催化材料均具有宽光谱吸收和高载流子分离效率的特点,为探索和设计新型高效极性光催化材料提供了一个全新的材料体系。
3.4 其 他
除了以上用于拓展光吸收和促进光生载流子分离的设计理论之外,作者团队还在光催化材料的晶面调控、复合材料、有机-无机复合光催化材料等方面进行了大量研究,提出了包括“晶面间电荷转移理论”、“价态异质结”、“光热协同光催化”等新型高效光催化材料的设计理论,并探索了一系列新型光催化材料,大大拓展了光催化材料的材料体系[34-41]。
4 结 语
光催化技术作为解决人类社会能源短缺和环境污染问题的最有效解决方案之一,成为近期世界范围内的研究热点之一。然而,较低的光催化转化效率,依然严重阻碍了光催化技术的进一步发展及其广泛应用。因此,进一步提高光催化材料的转化效率是未来光催化技术领域所面临的关键问题。虽然,人们在过去的几十年时间里为提高光催化效率进行了大量的研究工作。但是,目前较窄的光吸收范围和较低的载流子分离效率依然是制约光催化材料催化活性的最关键问题。如何进一步拓展光催化材料的光吸收范围,并同时提高光生载流子的分离效率,是设计和探索新型高效光催化材料的关键。
不同于拓展光催化材料光吸收范围和提高其载流子分离的常规手段,我们从材料的晶体结构出发,通过晶体结构和电子结构设计,提出了一些新的光催化材料设计理论,如:表面等离子体光催化材料、离子间电荷转移、极性内建电场促进光生载流子分离等,并制备了一系列具有宽光谱吸收和高载流子分离效率的新型高效光催化材料。本文主要根据近期作者课题组在新型光催化材料设计与制备方面的研究进展,提出一些关于材料设计理论方面的一些见解和看法,希望对从事光催化技术研究的广大同行能够有所启发和帮助。相信在国家的大力支持和广大同行的共同努力下,我国光催化技术一定能够在将来得到更好的发展和广泛的应用,为解决我国能源和环境问题作出贡献。
References
[1] Wang Peng, Huang Baibiao, Zhang Xiaoyang,etal.AngewChemIntEd[J], 2008, 47: 7931-7933.
[2] Wang Peng, Huang Baibiao, Zhang Xiaoyang,etal.ChemEurJ[J], 2009, 15: 1821-1823.
[3] Wang Peng, Huang Baibiao, Zhang Qianqian,etal.ChemEurJ[J], 2010, 16: 10042-10047
[4] Wang Peng, Huang Baibiao, Zhang Xiaoyang,etal.ChemCatChem[J], 2011, 3: 360-364.
[5] Wang Peng, Huang Baibiao, Lou Zaizhu,etal.ChemEurJ[J], 2010, 16: 538-544.
[6] Wang Peng, Huang Baibiao, Qin Xiaoyan,etal.InorgChem[J], 2009, 48: 10697-10702.
[7] Cheng Hefeng, Huang Baibiao, Wang Peng,etal.ChemCommun[J], 2011, 47: 7054-7056.
[8] Cheng Hefeng, Huang Baibiao, Dai Ying,etal.Langmuir[J], 2010, 26, 6618-6624.
[9] Cheng Hefeng, Huang Baibiao, Wang Junpeng,etal.JMaterChemA[J], 2013, 1: 7131-7136
[10]Lou Zaizhu, Huang Baibiao, Qin Xiaoyan,etal.CrystEngComm[J], 2011, 13: 1789-1793.
[11]Lou Zaizhu, Huang Baibiao, Ma Xiangchao,etal.ChemEurJ[J], 2012, 18: 16090-16096.
[12]Lou Zaizhu, Huang Baibiao, Qin Xiaoyan,etal.ChemCommun[J], 2012, 48: 3488-3490.
[13]Lou Zaizhu, Huang Baibiao, Wang Zeyan,etal.DaltonTrans[J], 2013, 42: 15219-15225.
[14]Lou Zaizhu, Huang Baibiao, Wang Peng,etal.DaltonTrans[J], 2011, 40: 4104-4110.
[15]Zheng Zhaoke, Huang Baibiao, Qin Xiaoyan,etal.JMaterChem[J], 2011, 21: 9079-9087.
[16]Ma Xiangchao, Dai Ying, Gao Meng,etal.PhysChemChemPhys[J], 2013, 15: 8722-8731.
[17]Ma Xiangchao, Dai Ying, Yu Lin,etal.JPhysChemC[J], 2014, 118: 12133-12140.
[18]Ma Xiangchao, Dai Ying, Huang Baibiao.ACSApplMaterInterf[J], 2014, 6: 12388-12394.
[19]Ma Xiangchao, Dai Ying, Yu Lin,etal.Light:Science&Applications[J], 2016, 5: e16017.
[20]Ma Xiangchao, Dai Ying, Yu Lin,etal.SciRep[J], 2014, 4: 3986.
[21]Cheng Hefeng, Huang Baibiao, Hiromi Yamashita,etal.JAmChemSoc[J], 2016, 138 (29): 9316-9324.
[22]Wang Gang, Huang Baibiao, Ma Xiangchao,etal.AngewChemIntEd[J], 2013, 52: 4810-4813.
[23]Li zhujie, Dai Ying, Ma Xiangchao,etal.PhysChemChemPhys[J], 2014, 16: 3267.
[24]Wang Gang, Huang Baibiao, Li Zhujie,etal.ChemEurJ[J], 2015, 21: 13583-13587.
[25]Wang Wenjun, Huang Baibiao, Ma Xiangchao,etal.ChemistryaEuropeanJournal[J], 2013 (19): 14777-14780.
[26]Zhang Rui, Dai Ying, Huang Baibiao,etal.CrystEngComm[J], 2014, 16: 4931-4934.
[27]Jiang Zaiyong, Liu Yuanyuan, Huang Baibiao,etal.SciRep[J], 2016, 6: 22727.
[28]Wang Wenjun, Cheng Hefeng, Huang Baibiao,etal.InorgChem[J], 2014, 53: 4989-4993.
[29]Zhang Bo, Wang Zeyan, Huang Baibiao,etal.ChemMater[J], 2016, 28: 6613-6620.
[30]Lou Zaizhu, Huang Baibiao, Wang Zeyan,etal. ChemMater[J], 2014, 26: 3873-3875.
[31]Lou Zaizhu, Wang Zeyan, Huang Baibiao,etal.ChemEurJ[J], 2015, 21: 8706-8710.
[32]Zhu Xianglin, Wang Zeyan, Huang Baibiao,etal.APLMater[J], 2015, 3: 104413.
[33]Zhu Xianglin, Wang Peng, Huang Baibiao,etal.AppliedCatalysisB:Environmental[J], 2016, 199: 315-322.
[34]Zheng Zhaoke, Huang Baibiao, Wang Zeyan,etal.JPhysChemC[J], 2009, 113: 14448-14453.
[35]Zheng Zhaoke, Huang Baibiao, Dai Ying,etal.ChemEurJ[J], 2011, 17: 15032-15038.
[36]Wang Gang, Huang Baibiao, Wang Lu,etal.ChemCommun[J], 2014, 50: 3814-3816.
[37]Wang Gang, Huang Baibiao, Luo Zaizhu,etal.AppliedCatalysisB:Environmental[J], 2016, 180: 6-12.
[38]Wang Guanzhi, Sun Qilong, Liu Yuanyuan,etal.ChemEurJ[J], 2015, 21: 2364-2367.
[39]Wang Guanzhi, Liu Yuanyuan, Huang Baibiao,etal.DaltonTrans[J], 2015, 44: 16238-16241.
[40]An Yang, Liu Yuanyuan, Huang Baibiao,etal.JSolidStateChem[J], 2016, 233: 194-198.
[41]Liu Yuanyuan, Wang Guanzhi, Huang Baibiao,etal.JCollidInterfSci[J], 2016, 469: 231-236.
(编辑 盖少飞)
Design and Synthesis of Highly Reactive Photocatalysts
WANG Zeyan1, HUANG Baibiao1, DAI Ying2
(1.State Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, China) (2.School of Physics, Shandong University, Jinan 250100, China)
Photocatalysis is regarded as one of the most promising strategies to solve energy and environmental problems of human society, which has attracted increasing interests all over the world. However, the further development and practical applications are greatly limited owing to the low photocatalytic efficiencies. Therefore, it is the most important issue to design and synthesize new kinds of photocatalysts with high conversion efficiencies. We recently proposed some new ideas on designing and synthesis of highly reactive photocatalysts from the aspect of crystal structure design, which have been summarized in this work in details. In this work, we mainly focused and presented some information on the designing theories, synthesis methods, structure-property relationship, and photocatalytic mechanism of several new photocatalysts including plasmonic photocatalysts, infrared photocatalysts and polar-photocatalysts.
photocatalysis; light absorption; photogenerated charge separation; design and synthesis of materials
2016-09-30
科技部“973”计划项目(2013CB632401);国家自然科学基金资助项目(21333006)
王泽岩,男,1981年生,副教授,硕士生导师
黄柏标,男,1962年生,教授,博士生导师,Email: bbhuang@sdu.edu.cn
10.7502/j.issn.1674-3962.2017.01.02
O643.36;O644.1
A
1674-3962(2017)01-0007-10
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
物理学报(2022年6期)2022-03-30
石油化工高等学校学报(2021年3期)2021-07-15
无机盐工业(2020年1期)2020-12-31
陕西科技大学学报(2020年6期)2020-11-25
人工晶体学报(2020年9期)2020-10-21
物理学报(2020年16期)2020-08-29
无线互联科技(2019年15期)2019-11-07
物理学报(2019年16期)2019-08-29
