
基于氯化血红素复合材料的过氧亚硝酸阴离子电化学传感器的构建
2017-02-06 21:27任聚杰于聪聪崔敏李亚清武聪籍雪
分析化学 2017年1期
关键词:碳纳米管
任聚杰+于聪聪+崔敏+李亚清+武聪+籍雪平

摘 要 构建了一种基于氯化血红素/金纳米粒子/聚三聚氰胺/多壁碳纳米管复合材料修饰玻碳电极的过氧亚硝酸阴离子电化学传感器,并成功用于过氧亚硝酸阴离子的检测。采用循环伏安法和电流.时间曲线考察了过氧亚硝酸阴离子在传感器上的电化学行为,并对传感器的制备条件及过氧亚硝酸阴离子的检测条件进行了优化。结果表明,碳纳米管滴涂量为5 μL,金沉积时间为20 s,工作电位为0.8 V时,所制传感器对过氧亚硝酸阴离子的响应最大。在优化实验条件下,此传感器检测过氧亚硝酸阴离子的线性范围为1.0×105~3.5×104 mol/L和3.5×104~1.1×103 mol/L,灵敏度为0.13 A/(mol/L),检出限为1.2×107 mol/L(S/N=3)。
关键词 过氧亚硝酸阴离子; 氯化血红素; 聚三聚氰胺; 金纳米粒子; 碳纳米管; 电化学传感器
1 引 言
在有氧代谢的过程中,生物体会产生多种自由基,活性氮[1](RNS)是其中重要的一类,它主要是指一氧化氮(NO)与包括活性氧(ROS)在内的化合物相互作用,衍生出一系列具有高度氧化活性的自由基和硝基类化合物,包括过氧亚硝酸阴离子(ONOO)、一氧化氮(NO)、亚硝酸根离子(NO-2)和二氧化氮(NO2)等。在生物体内,ONOO由一氧化氮和超氧阴离子两种自由基快速结合产生,一氧化氮和超氧阴离子本身都不是强氧化剂,但生成的ONOO却具有极强的氧化性和硝化性[2,3]。ONOO
能氧化巯基蛋白和脂质、诱导单链DNA的断裂、引起一些DNA或蛋白质的氨基酸残基的硝化[4~7]等,参与诸多疾病的病理过程,主要包括心血管疾病、癌症、糖尿病和神经退行性疾病等。因此,准确定量检测ONOO在疾病的早期诊断和治疗方面具有重要意义[8,9]。
目前,检测ONOO的方法主要有紫外分光光度法[10]、荧光法[11~13]、化学发光法[14,15]、电子自旋共振光谱法[16,17]、电子自旋与液相色谱.质谱联用法[18]和电化学方法[19~24]等。与电化学方法相比,其它方法存在着一些不足,如电子自旋共振法或与其它技术联用的方法只是对ONOO氧化生物活性物质的机理进行了一些研究; 荧光法则是通过合成荧光探针间接地测定ONOO,但是有些探针的合成比较困难,成本较高; 而电化学方法操作简单,灵敏度高,选择性好,成本低。利用电化学方法检测ONOO的报道较少,2007年, Cortés等采用聚锰酞菁膜修饰的铂微电极实现了对ONOO的实时安培检测[19],但是电极表面的电荷转移可能会受pH值的影响。Wang等[8]采用聚氰钴氨膜修饰的玻碳电极成功构建了一种ONOO电化学传感器,并实现了人体血清样品中ONOO的检测,但灵敏度有待提高。近几年,Peteu等利用导电聚合物聚3,4.乙烯二氧噻吩(PEDOT)或者石墨烯复合材料构建了几种过氧亚硝酸阴离子电化学传感器 [21~24],并且对ONOO的检测能达到较低的检出限,但未给出工作曲线。氯化血红素是人工合成的一种具有π共轭结构的卟啉分子,它能模拟超氧化物歧化酶与ONOO反应 [9,18]。多壁碳纳米管具有离域的大π键结构,使得多壁碳纳米管与卟啉分子之间存在强烈的π.π相互作用[25],本研究利用这种相互作用将氯化血红素固定到多壁碳纳米管修饰的电极表面,同时修饰具有特殊形貌的金纳米粒子来提高传感器的灵敏度,构建了一种新型的过氧亚硝酸阴离子电化学传感器。
2 实验部分
2.1 仪器与试剂
CHI660D电化学工作站(上海辰华仪器有限公司),采用传统的三电极体系:直径为3 mm的玻碳电极(Glassy carbon electrode, GCE)或修饰的玻碳电极作为工作电极,饱和银.氯化银电极为参比电极,铂片电极为对电极; BT125D电子分析天平(北京赛多利斯科学仪器有限公司); UV5200.PC紫外可见分光光度计(上海元析仪器有限公司); S.4800冷场发射扫描电子显微镜(SEM, 日本日立公司); 石英自动双重纯水蒸馏器(江苏金坛宏华仪器厂); 雷磁PHS.2F型pH计(雷磁.上海仪电科学仪器股份有限公司); 移液枪10 μL、100 μL(北京青云卓立精密仪器有限公司)。
多壁碳纳米管(Multiple.walled carbon nanotubes, MWCNTs,深圳市纳米港有限公司); Na2HPO4、N,N.二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、MnO2(国药集团化学试剂有限公司); 氯化血红素(Hemin)、三聚氰胺(Melamine, Mel)、四氯金酸(HAuCl4·3H2O)、H2O2(30%,w/w)、NaOH、NaNO2(上海晶纯生化科技股份有限公司); 其它试剂均为分析纯; 实验用水均为二次蒸馏水,所有实验都是在氮气氛围下进行的。
2.2 部分所需溶液的配制
称取多壁碳纳米管1.5 mg,溶于1 mL DMF中,超声分散30 min,得到分散均匀的MWCNTs悬浮液。配制0.05 mol/L Na2HPO4溶液,并用NaOH调节至pH 11,作为PBS溶液。实验中的过氧亚硝酸阴离子储备液(Peroxynitrite, PON)采用文献[26]中的方法制备,制得的PON于18℃保存。在电化学测试前,采用紫外分光光度计在λ=302 nm下测其吸光度,利用朗伯.比尔定律,计算PON浓度。
2.3 修饰电极的制备
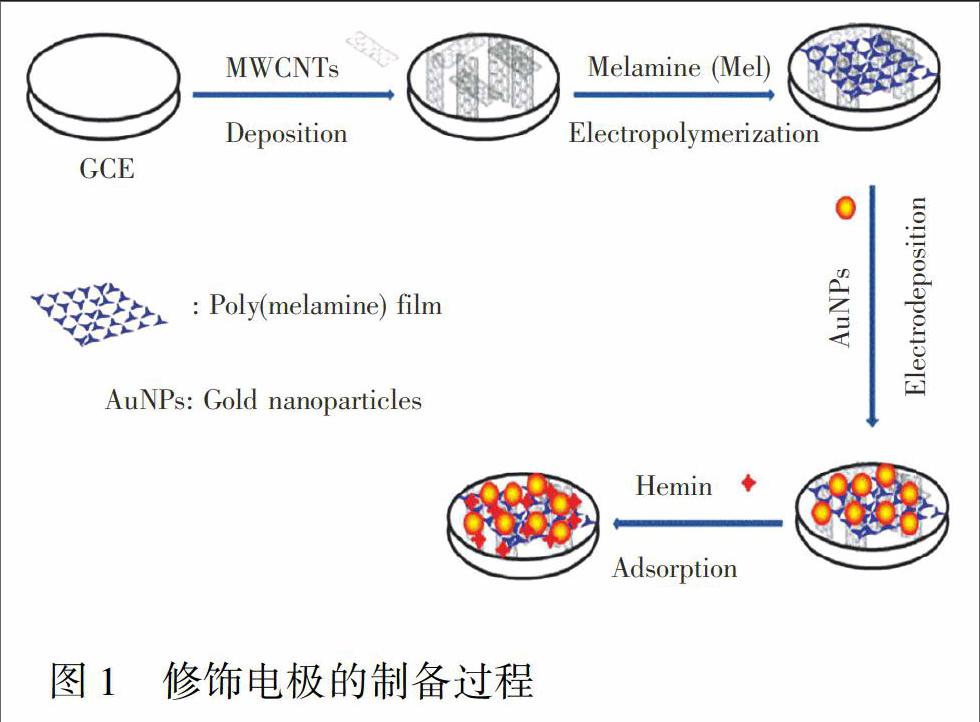
本研究以玻碳电极为基础电极,图1为修饰电极的制备过程图。首先,玻碳电极依次采用1.0,0.3和0.05 μm的三氧化二铝粉末在抛光板上打磨,每次打磨后依次在水、无水乙醇、水中超声清洗,并用氮气吹干。
用移液枪吸取MWCNTs悬浮液5 μL,滴涂到预处理好的玻碳电极上,过夜晾干待用,作为MWCNTs/GCE。将得到的MWCNTs/GCE电极置于含1.00 mmol/L Mel的0.1 mol/L HCl溶液中,采用循环伏安法电聚合10圈,电位区间为0.2~1.5 V,扫速为50 mV/s,取出后用大量水冲洗,以去除电极表面未聚合的三聚氰胺,室温下以氮气吹干待用,作为PMel/MWCNTs/GCE。将PMel/MWCNTs/GCE电极插入到1.00 mmol/L HAuCl4+0.5 mol/L H2SO4溶液中,恒电位
0.2 V下电沉积20 s,得到的电极用水冲洗干净,氮气吹干,作为AuNPs/PMel/MWCNTs/GCE。将电极AuNPs/PMel/MWCNTs/GCE插入到含1.00 mmol/L Hemin的DMSO溶液中浸泡15 h,得到的电极用大量水冲洗干净并用氮气吹干,作为Hemin/AuNPs/PMel/MWCNTs/GCE。
2.4 电化学测量
在测定PON时,采用循环伏安法(Cyclic voltammetry, CV)在PBS溶液中扫描,电位区间为0.4~1.2 V,扫速为50 mV/s。电化学交流阻抗(Electrochemical impedance spectroscopy, EIS)在[Fe(CN)6]34(含0.1 mol/L KCl)溶液中进行,频率范围为0.1~100000 Hz, 振幅为5 mV。电流.时间曲线(Amperometric i.t Curve, i.t)在0.05 mol/L PBS溶液中进行,并采用磁力搅拌仪恒速微搅拌。
3 结果与讨论
3.1 修饰电极的电化学行为
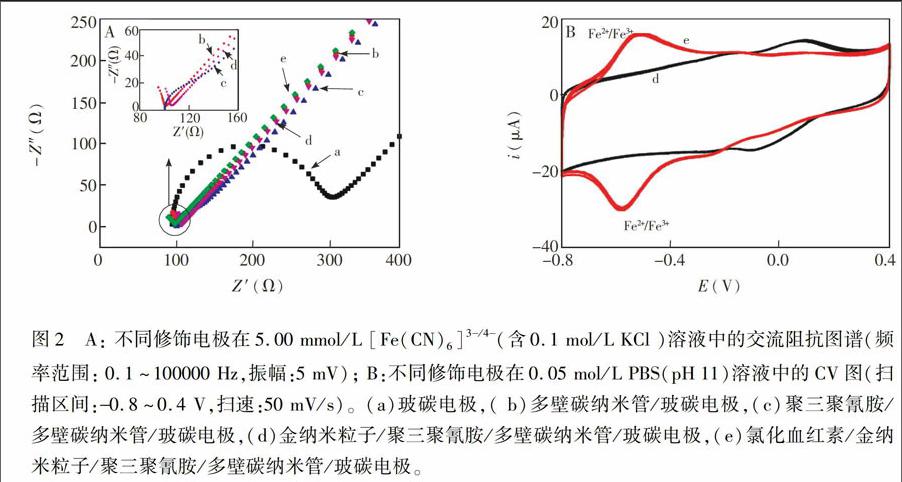
电化学交流阻抗(EIS)是研究电极过程动力学、电极表面现象等的重要手段[27] 。在实验中,采用EIS表征各阶段修饰电极的电化学性能。由图2A可见,裸电极GCE(曲线a)的电子转移电阻(Rct)约为200 Ω,修饰了碳纳米管后的MWCNTs/GCE电极(曲线b)的Rct值几乎为零,这是因为碳纳米管具有优良的导电性能,同时也说明碳纳米管成功修饰到了电极表面; 由插图可以看出,再修饰聚三聚氰胺得到的PMel/MWCNTs/GCE电极(曲线c)的Rct又稍有增大,推测这是因为聚三聚氰胺的导电性不及碳纳米管,同时也说明聚三聚氰胺成功修饰到了电极表面; 再沉积金得到的AuNPs/PMel/MWCNTs/GCE电极(曲线d)的Rct值又几乎为零,说明金纳米粒子成功修饰到电极上,并且电极上修饰的纳米复合材料AuNPs/PMel/MWCNTs的导电性非常好,它们之间的协同作用共同促进了电子传递; 修饰Hemin后得到的Hemin/AuNPs/PMel/MWCNTs/GCE电极(曲线e)的Rct值再无明显变化,说明最终的修饰电极的阻抗较小,有利于电子在电极与电解液之间的传递。
图2B为电极AuNPs/PMel/MWCNTs/GCE(曲线d)和电极Hemin/AuNPs/PMel/MWCNTs/GCE(曲线e)在PBS溶液里的循环伏安曲线,扫描圈数为3圈,Hemin/AuNPs/PMel/MWCNTs/GCE(曲线e)电极在0.6 V附近出现了一对可逆的氧化还原峰,这是Hemin中Fe3+/Fe2+的峰,与文献[28,29]报道一致,进一步说明Hemin成功修饰到了电极上。
3.2 修饰电极的SEM表征
采用扫描电子显微镜对不同修饰电极表面的形态进行表征。由图3可见,Hemin/AuNPs/MWCNTs/GCE电极(图3a)上的金纳米粒子分散不均匀,出现了严重的团聚现象; 修饰有聚三聚氰胺的电极Hemin/AuNPs/PMel/MWCNTs/GCE(图3b)上的金纳米粒子分散比较均匀; 金纳米粒子形成了很多小的粒子簇,具有特殊的花朵形貌(图3c),推测这是由于聚三聚氰胺对碳纳米管的修饰,改善了金纳米粒子在碳纳米管上的分散状况,与文献[30,31]报道的氨基修饰的碳材料在预防纳米粒子团聚方面是有帮助的说法一致。
3.3 PON在不同修饰电极的电化学行为
图4为0.7 mmol/L PON在不同修饰电极上的循环伏安(CV)曲线。由图可知,CV曲线只出现氧化峰,无还原峰,说明在此电位范围内PON的氧化是一个不可逆过程,与文献[8]报道的ONOO的氧化是不可逆的一致。在GCE(曲线a)上,PON的氧化峰电位约在1.0 V,且峰形较缓,电流较小。在Hemin/MWCNTs/GCE(曲线b)上,PON的氧化电位负移了约150 mV,峰电流增大约1.5倍,这可能是因为碳纳米管具有良好的导电性,Hemin对PON具有选择催化作用,两者的修饰促进了电子传递的速率; 在Hemin/PMel/MWCNTs/GCE(曲线c)上,PON的氧化电流值相对于GCE几乎无明显变化,但是在修饰有金纳米粒子的Hemin/AuNPs/MWCNTs/GCE(曲线d)和Hemin/AuNPs/PMel/MWCNTs/GCE(曲线e)上,PON的氧化峰电流相对于GCE分别增大了2倍和3倍,这说明金纳米粒子的修饰,可以促进PON在电极表面的氧化,使得PON的氧化峰电流增大,并且在修饰了聚三聚氰胺的电极上PON的氧化峰电流更大,推测这是因为修饰有聚三聚氰胺的电极表面的金纳米粒子分散比较均匀,使得Hemin/AuNPs/PMel/MWCNTs/GCE的比表面积更大,因此,对PON的催化效果更好。
3.4 实验条件的优化
对传感器的制备条件进行了优化,由图5A可见, 金沉积时间对Hemin/AuNPs/PMel/MWCNTs/GCE的影响,当金沉积时间小于20 s时,电极对PON的循环伏安峰电流随着金沉积时间的延长而增大,金沉积20 s时,峰电流达到最大值; 当金沉积时间大于20 s后,电极对PON的循环伏安峰电流随金沉积时间的延长而减小。因此,最佳金沉积时间为20 s。
由图5B可见,不同的碳纳米管滴涂量对Hemin/AuNPs/PMel/MWCNTs/GCE的影响,当碳纳米管的滴涂量小于5 μL时,电极对PON的电流响应随碳纳米管滴涂量的增加而增大; 滴涂量为5 μL时峰电流达到最大值; 当滴涂量大于5 μL后,电极对PON的电流响应随碳纳米管滴涂量的增加而减小。因此,最佳碳纳米管滴涂量为5 μL。
3.5 传感器对PON的响应
采用电流.时间曲线,研究了Hemin/AuNPs/PMel/MWCNTs/GCE对PON的催化性能。
如图7所示,在0.80 V的工作电位下,向PBS溶液中连续加入PON溶液,结果表明,PON浓度在1.0×105~ 3.5×104 mol/L以及3.5×104~ 1.1×103 mol/L区间时,PON的电流与其浓度均满足线性关系,对应的线性方程分别为I1(μA)=0.13C(μmol/L)-0.714(R2=0.994)和I2(μA)=0.074C(μmol/L)+20.66(R2=0.990),如图7B所示。此传感器的灵敏度可达0.13 A/(mol/L),计算得到的检出限为1.2×107 mol/L(S/N=3),与其它修饰方法的比较结果见表1。由表1可知,本方法制得的传感器具有较宽的线性范围、较低的检出限和较高的灵敏度。
3.6 传感器的抗干扰性测定
3.7 电极的重现性和稳定性测定
采用相同的实验条件下制备的修饰电极,在0.05 mol/L PBS(pH 11)溶液中对0.7 mmol/L PON进行循环伏安测定5次,计算PON的峰电流的标准偏差为3.6%,说明制得的传感器有良好的重现性。对修饰电极的稳定性进行了测定,结果表明,将传感器在4℃保存两天后,在同样的实验条件下,再次测得电极对0.7 mmol/L PON的循环伏安电流响应值为初始电流的80%。
4 结 论
构建了一种基于氯化血红素复合材料修饰玻碳电极的过氧亚硝酸阴离子电化学传感器,结果表明,制备的传感器灵敏度高,检出限低,线性范围较宽,抗干扰性好,成本低,为PON的检测提供了一种新方法。
References
1 JING Xiao.Tong, YU Fa.Biao, CHEN Ling.Xin. Prog. Chem., 2014, 26(5): 866-878
景晓彤, 于法标, 陈令新. 化学进展, 2014, 26(5): 866-878
2 ZHENG Rong.Liang, HUANG Zhong.Yang. Free Radical Biology, Beijing: Higher Education Press, 2007: 33
郑荣梁, 黄中洋. 自由基生物学, 北京: 高等教育出版社, 2007: 33
3 Peteu S F, Boukherroub R, Szunerits S. Biosens. Bioelectron., 2014, 58(16): 359-373
4 Ohshima H, Tatemichi M, Sawa T. Arch. Biochem. Biophys., 2003, 417(1): 3-11
5 Deeb R S, Resnick M J, Mittar D, Mccaffrey T, Hajjar D P, Upmacis R K. J. Lipid. Res., 2002, 43(10): 1718-1726
6 Van′t Hof R J, Ralston S H. Immunology, 2001, 11(3): 255-261
7 Torreilles F, Salman.Tabcheh S, Guérin M C, Torreilles J. Brain Res. Rev., 1999, 30(2): 153-163
8 Wang Y, Chen Z Z. Talanta, 2010, 82(2): 534-539
9 Peteu S F, Bose T, Bayachou M. Anal. Chim. Acta, 2013, 780(10): 81-88
10 Vandervliet A, Eiserich J P, Oneill C A, Halliwell B, Cross C E. Arch. Biochem. Biophys., 1995, 319(2): 341-349
11 Wang B S, Yu F B, Li P, Sun X F, Han K L. Dyes Pigments, 2013, 96(2): 383-390
12 Ma J J, Wu J S, Liu W M, Wang P F, Fan Z Y. Spectrochim. Acta. A., 2012, 94(8): 340-345
13 Jia X T, Chen Q Q, Yang Y F, Tang Y, Wang R, Xu Y F, Zhu W P, Qian X H. J. Am. Chem. Soc., 2016, 138(34): 10778-10781
14 Dai K, Vlessidis A G, Evmiridis N P. Talanta, 2003, 59(1): 55-65
15 Adegoke O, Nyokong T. J. Lumin., 2013, 134(2): 448-455
16 Dikalov S, Skatchkov M, Bassenge E. Biochem. Bioph. Res. Co., 1997, 230(1): 54-57
17 Gielis J F, Boulet G A, Briedé J J, Horemans T, Debergh T, Kussé M, Cos P, van Schil P E Y. Eur. J. Cardio.Thorac., 2015, 48(4): 622-629
18 Imaram W, Gersch C, Kim K M, Johnson R J, Henderson G N, Angerhofer A. Free Radical. Bio. Med., 2010, 49(2): 275-281
19 Cortés J S, Granados S G, Ordaz A A, López Jiménez J A, Griveau S, Bedioui F. Electroanal, 2007, 19(1): 61-64
20 Quinton D, Griveau S, Bedioui F. Electrochem. Commun., 2010, 12(10): 1446-1449
21 Peteu S, Peiris P, Gebremichael E, Bayachou M. Biosens. Bioelectron., 2010, 25(8): 1914-1921
22 Koh W C, Son J I, Choe E S, Shim Y B. Anal. Chem., 2010, 82(24): 10075-10082
23 Oprea R, Peteu S F, Subramanian P, Wang Q, Pichonat E, Happy H, Bayachou M, Boukherroub R, Szunerits S. Analyst, 2013, 138(15): 4345-4352
24 Peteu S F, Whitman B W, Galligan J J, Swain G M. Analyst, 2016, 141(5): 1796-1806
25 Cheng F Y, Adronov A. Chem.Eur. J., 2006, 12(19): 5053-5059
26 Beckman J S, Chen J, Ischiropoulos H, Crow J P. Method. Enzymol., 1994, 233(23): 229-240
27 LI Qi.Long, HU Jin.Bo. Electroanalytical Chemistry. Beijing: Beijing Normal University Press, 2007: 444
李启隆, 胡劲波. 电分析化学, 北京: 北京师范大学出版社, 2007: 444
28 HUANG Xiu.Ling, WANG Qi.Hui, LIU Hui.Hong. J. Analyt. Sci., 2008, 24(1): 55-58
黄秀玲, 王启会, 刘慧宏. 分析科学学报, 2008, 24(1): 55-58
29 Guo Y J, Deng L, Li J, Guo S J, Wang E K, Dong S J. Acs Nano, 2011, 5(2): 1282-1290
30 Singh S K, Singh M K, Kulkarni P P, Sonkar V K, Grácio J J A, Dash D . Acs Nano, 2012, 6(3): 2731-2740
31 Lipińska M E, Rebelo S L H, Pereira M F R, Gomes J A N F, Freire C, Figueiredo J L. Carbon, 2012, 50(9): 3280-3294
Abstract A peroxynitrite electrochemical sensor based on hemin/gold nanoparticles/poly(melamine)/multi.walled carbon nanotube composite modifying glassy carbon electrode was successfully constructed for the detection of peroxynitrite. The electrochemical behavior of peroxynitrite on the sensor was investigated by cyclic voltammetry and amperometric i.t curve, and the conditions for preparation of the sensor and detection of peroxynitrite were optimized. The results showed that the amperometric response of the sensor for peroxynitrite reached the maximum when the amount of carbon nanotubes was 5 μL, the deposition time of gold was 20 s, and the working potential was 0.8 V. Under the optimized experimental conditions, the sensor could detect peroxynitrite in a linear range from 1.0×105 mol/L to 3.5×104 mol/L and from 3.5×104 mol/L to 1.1×103 mol/L, with a sensitivity of 0.13 A/(mol/L) and a detection limit of 1.2×107 mol/L (S/N=3).
Keywords Peroxynitrite; Hemin; Poly(melamine); Gold nanoparticles; Multi.walled carbon nanotubes; Electrochemical sensor
猜你喜欢
科学导报(2021年62期)2021-09-26
现代职业教育·中职中专(2018年12期)2018-06-11
有色金属材料与工程(2016年6期)2017-05-31
中国纤检(2016年12期)2017-01-20
百科知识(2016年16期)2016-10-29
科学导报(2016年62期)2016-07-09
新材料产业(2014年9期)2014-04-23
大科技·百科新说(2014年2期)2014-03-03
物理(2009年10期)2009-12-23
国外科技新书评介(2009年4期)2009-05-31
