
Leptin influences estrogen metabolism and increases DNA adduct formation in breast cancer cells
2017-01-13 01:54SamiaShoumanMohamedWagihMarwaKamel
Cancer Biology & Medicine 2016年4期
Samia Shouman, Mohamed Wagih, Marwa Kamel
1Department of Cancer Biology, Unit of Pharmacology, National Cancer Institute, Cairo University, Cairo 11796, Egypt;
2Department of Pathology, Faculty of Medicine, Beni-Suef University, Benisuef 62511, Egypt
Leptin influences estrogen metabolism and increases DNA adduct formation in breast cancer cells
Samia Shouman1, Mohamed Wagih2, Marwa Kamel1
1Department of Cancer Biology, Unit of Pharmacology, National Cancer Institute, Cairo University, Cairo 11796, Egypt;
2Department of Pathology, Faculty of Medicine, Beni-Suef University, Benisuef 62511, Egypt
Objective: The elevated incidence of obesity has been paralleled with higher risks of breast cancer. High adiposity increases leptin secretion from adipose tissue, which in turn increases cancer cell proliferation. The interplay between leptin and estrogen is one of the mechanisms through which leptin influences breast carcinogenesis. An unbalanced estrogen metabolism increases the formations of catechol estrogen quinones, DNA adducts, and cancer mutations. This study aims to investigate the effect of leptin on some estrogen metabolic enzymes and DNA adduction in breast cancer cells.
Methods: High performance liquid chromatography (HPLC) was performed to analyze the DNA adducts 4-OHE1[E2]-1-N3 adenine and 4-OHE1[E2]-1-N7 guanine. Reporter gene assay, real time reverse transcription polymerase chain reaction (real time RT-PCR), and Western blot were used to assess the expression of estrogen metabolizing genes and enzymes: Cytochrome P-450 1B1 (CYP1B1), Nicotinamide adenine dinucleotide phosphate-quinone oxidoreductase1 (NQO1), and Catechol-O-methyl transferase (COMT).
Results: Leptin significantly increased the DNA adducts 4-OHE1[E2]-1-N3 adenine and 4-OHE1[E2]-1-N7 guanine. Furthermore, leptin significantly upregulated CYP1B1 promoter activity and protein expression. The luciferase promoter activities of NQO1 and mRNA levels were significantly reduced. Moreover, leptin greatly reduced the reporter activities of the COMT-P1 and COMT-P2 promoters and diminished the protein expression of COMT.
Conclusions: Leptin increases DNA adduct levels in breast cancer cells partly by affecting key genes and enzymes involved in estrogen metabolism. Thus, increased focus should be directed toward leptin and its effects on the estrogen metabolic pathway as an effective approach against breast cancer.
Breast cancer; leptin; estrogen metabolism; DNA adducts
Introduction
Breast cancer, the most frequently occurring cancer among women, is a major public health problem. The worldwide incidence of female breast cancer is predicted to reach approximately 3.2 million new cases annually by 2050. This reflects the magnitude of breast cancer incidence and its effect on society worldwide, as well as the urgent need for preventive and treatment measures1.
Estrogen metabolites are considered as breast cancer risk factors. The estrogen metabolic pathway is regulated by the oxidizing phase 1 and conjugating phase 2 enzymes. Cytochrome P-450 1A1 (CYP1A1) and cytochrome P-450 1B1 (CYP1B1) catalyze the oxidation of estrone (E1) or 17-β estradiol (E2) to catechol estrogens 2-OHE1[E2] and 4-OHE1[E2], respectively. These catechol estrogens can be detoxified by Catechol-O-methyl transferase (COMT) to form methoxyestrogens 2-MeOE1[E2] and 4-MeOE1[E2], or they may be further oxidized by CYPs to semiquinones (SQ) and quinones (Q), accordingly. Estrogen quinones may be detoxified through reduction by nicotinamide adenine dinucleotide phosphate-quinone oxidoreductase1 (NQO1), or they may form quinone-DNA adducts or oxidative DNA adducts through quinone-semiquinone redox cycling2,3(Figure 1). Depurinating adducts are formed when carcinogens covalently bind at the N3 or N7 of adenine or the N7 of guanine. Quinones from 4-OHE1[E2] and to a much lesser extent from 2-OHE1[E2] react with DNA and generate critical mutations4,5. Moreover, stable bulky guanine adducts of 4-OHE1[E2] have been detected in human breast tumor tissue6, and unbalanced estrogen metabolism has been implicated in breast cancers7. Consequently, research on compounds that affect the estrogen metabolic pathway isongoing. Example of these compounds are the dietary supplements resveratrol and N-acetylcysteine8and the tumor necrosis factor-alpha (TNF-α)9, as well as vegetable and carbohydrate consumption10.
Obesity is another vital risk for breast cancer in postmenopausal women, which might involve interlinked molecular mechanisms in the pathogenesis. Increased levels of estrogens caused by the aromatization of adipose tissues, inflammatory cytokines, such as TNF-α, interleukin-6, and prostaglandin E2, insulin resistance, and hyperactivation of insulin-like growth factors pathways, adipokines, and oxidative stress are all aberrantly regulated in obese women as these undoubtedly contribute to carcinogenesis. These molecular factors interfere with intracellular signaling in the MAPK and mTOR pathways, which regulate the progression of cell cycle, apoptosis, and protein synthesis. In this context, the structural defects of typical genes related to both breast cancer and obesity, such as leptin, leptin receptors, serum paraoxonase/arylesterase-1, fat mass and obesity associated gene, and melanocortin receptor 4 have been associated with varying risks of breast cancer development. Accordingly, the early detection of these gene alterations can be used as risk predictors in obese women, and targeting these pathways involved in the breast cancer pathogenesis in obese women is also a potential therapeutic tool11.
Thus, the role of leptin in the core of obesity is significant given that adipose tissues are the main site of leptin secretion. Breast cancers in obese individuals show increased inflammation, hypoxia, angiogenesis, and changes in endocrine factors including insulin, IGF-1, leptin, adiponectin, and estrogen12. Obese tissue adipocytes upregulate chemokines and leptin which attract macrophages that produce proinflammatory proteins (TNF-α and IL1b) and proangiogenic factors, providing a microenvironment conducive for adipose tissue expansion and tumor progression13. High leptin levels in obese patients may exaggerate the inflammatory status that disposes patients to later distant metastasis14. Recently, leptin was found to play a role in metabolic reprogramming that involves the enhanced use of glucose for biosynthesis and lipids for energy production. These metabolic adaptations induced by leptin may benefit the MCF-7 growth and support the reverse Warburg effect designated in breast cancer15. Moreover, studies report the existence of a crosstalk between leptin and estrogen, e.g., leptin can initiate the signaling pathways activated when estrogen receptor α (ERα) is simulated by estradiol. Leptin can also prompt the aromatase activity in MCF-7 breast cancer cells, enhance in situ estradiol production, and endorse estrogen dependent breast cancer proliferation. Furthermore, leptin can interfere with the anticancer effects of the antiestrogen ICI 182, 780, all of which elucidate the role of leptin in enhancing breast cancer growth16. Low levels of serum estradiol recently demonstrated that it may promote adipocyte genesis by influencing leptin function17. However, despite this interplay between leptin and estrogen in breast cancer, the relation between leptin and estrogen metabolism remains unexplored.
Thereby, this study is rooted in the hypothesis that specific estrogen metabolites react with DNA to form depurinating estrogen DNA adducts. The resulting apurinic sites in the DNA can generate critical mutations leading to breast cancer. Our work focuses on the effect of leptin on some important enzymes involved in the estrogen metabolic pathway which may affect the level of DNA adducts formation in breast cancer cells. We anticipate that this will enrich our understanding of the mechanisms linking obesity and breast cancer, and thus, offer significant opportunities to target and prevent this devastating disease.
Materials and methods
Cells and cell culture
The human breast cancer cell line MCF-7 was purchasedfrom Rockville culture collection, Md, USA. The cells were maintained at 37 °C in a 95% humidified, 5% CO2atmosphere in RPMI-1640 supplemented with 10% heat inactivated fetal bovine serum, 1% L-glutamine, 1% sodium pyruvate, and 1% penicillin and streptomycin (Gibco BRL, Life Technologies, Long Island, USA). To study the effect of leptin on the formation of DNA adducts, the cells were grown in a serum free phenol red free media for 24 h. The cells were then treated with a 10 nM of E2 with or without leptin for 48 h. The media were collected for DNA adducts analysis.
High performance liquid chromatography (HPLC) analysis of estrogen induced depurinating DNA adducts
Sample preparation
MCF-7 cells were treated as described above. Growth media were used to measure the level of depurinating DNA adducts according to the method described by Saeed et al.18. Cell culture media were extracted using Varian C8 Certify II solid phase extraction cartridges (Varian, Harbor City, USA), which were pre-equilibrated by sequentially passing 1 mL methanol, distilled water, and potassium phosphate buffer (100 mM, pH 8.0). Culture media was adjusted with 1 mL of 1 M potassium phosphate to pH 8.0 and passed through the cartridge. After washing with 2 mL of 100 mM potassium phosphate and 5 mL of distilled water, the analytes were eluted with 1 mL of elution buffer consisting of methanol/ acetonitrile/water/TFA (8:1:1:0.1). The eluant was evaporated to about 100 μL by using Jouan RC10 Vacuum Concentrator and reconstituted with 100 μL of methanol/water (1:1). Subsequently, the solution was passed through 5000 MW cut-off filters and analyzed on HPLC connected with multiple electrochemical detectors.
HPLC analysis of adducts
The analysis of all samples was performed on an HPLC system equipped with dual ESA Model 580 autosampler and a 12-channel CoulArray electrochemical detector (ESA, Chelmsford, USA). The two mobile phases used are as follows: acetonitrile/methanol/buffer/water (15:5:10:70) and acetonitrile/methanol/buffer/water (50:20:10:20). The buffer was a mixture of 0.25 M citric acid and 0.5 M ammonium acetate in triple distilled water, and the pH was adjusted to 3.6 with acetic acid. The 95 μL injections were carried out on a Phenomenex Luna-2 C-18 column (250 × 4.6 mm ID, 5 mm; Phenomenex, Torrance, USA), initially eluted isocartically at 90%A/10%B for 15 min followed by a linear gradient to 90%B/10%A in the next 40 min and retained for 5 min (total 50 min gradient) at a flow rate of 1 mL/minute and a temperature of 30 °C. The serial array of 12 coulometric electrodes was set at potentials of -35, 10, 70, 140, 210, 280, 350, 420, 490, 550, 620, and 690 mV. The system was controlled, and the data were processed using the CoulArray software package (ESA). Peaks were identified by both retention time and peak height ratios between the dominant peaks and the peaks at the two adjacent channels. The metabolites, conjugates, and depurinating adducts were quantified by comparing peak response ratios with known amounts of standards. The level of adducts were normalized against cell numbers and DNA contents.
Reporter assay
Stable transfection of MCF-7 with the reporter plasmids
Luciferase reporter plasmids containing CYP1B1, COMTP1 (proximal COMT-promoters), COMTP2 (distal COMT-promoters), and NQO1 promoters were used in this study. These plasmids were stably transfected into MCF-7 according to the following: MCF-7 cells were cotransfected with individual reporter plasmid (10 μg) and Neomycinexpressing vector (1 μg) using transfection reagent Fugene 6 (Roche, Indianapolis, USA) following the manufacturer's protocol. At 48 h post-transfection, the media were replaced with Genecitin containing media (500 μg/mL media). Individual colonies were picked and propagated following 2 weeks of selection and were screened for luciferase activity. Colonies which showed positive luciferase activity indicated that they were successfully stably transfected and thus were selected and maintained in liquid nitrogen for further experiments.
Luciferase assay
The stably transfected cells containing the reporter gene vectors with the promoters of interest were grown in RPMI media, distributed in 6 well plates, and treated with varying leptin concentrations. After 48 h, the cells were harvested, and luciferase activities were determined using luciferase enzyme assay systems according to the supplier’s protocol (Promega, Madison, USA). The luciferase activity was then normalized against protein concentration using the Bradford protein assay procedure.
Real time RT-PCR
MCF-7 cells were cultured, distributed in 6 well plates, and treated with varying leptin concentrations. After 48 h, total RNA was isolated through RNA aqueous-Micro kit (Ambion, Foster City, USA) according to the manufacturer’s instructions. cDNA synthesis was conducted using High Capacity cDNA Reverse Transcription kit (Applied Biosystem, Foster city, USA) following the manufacturer’s protocol. PCR amplification was performed using Taqman Fast Universal PCR Master Mix (Applied Biosystem, Foster city, USA). Quantitative real time RT-PCR was carried out using the predeveloped Taqman assay reagents control kits (Applied Biosystem, Foster city, USA). Kits were used for NQO1 (assay ID Hs00168547-ml), COMT (assay ID Hs00241349-ml) and glyceraldehyde-3-phosphate dehydrogenase (assay ID Hs9999905-ml). Each assay was performed in duplicate.
Western blot analysis
MCF-7 cells were cultured, distributed in 6 well plates, and treated with varying leptin concentrations. After 72 h, the cells were washed with phosphate buffered saline (PBS) and whole cell lysates were prepared with radioimmunopreciptation assay (RIPA) lysis buffer. Cell lysates were solubilized in a sample buffer [60 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 0.7 mol/L βmercaptoethanol; 0.01% bromophenol blue] and subjected to SDS polyacrylamide gel electrophoresis (PAGE). Proteins were electroblotted onto nitrocellulose membranes. Membranes were immunoblotted with the primary antibodies against CYP1B1 and COMT (Chemicon International Inc., Temecula, USA), NQO1 (Novus Biologicals Inc, Littleton, USA) and β-actin (Sigma-Aldrich Co., St. Louis, USA). After washing, membranes were incubated with horse radish peroxidase (HRP) conjugated secondary antibodies (Vector Laboratories, Burlingame, USA), and x-ray films were then developed.
Statistical analysis
Data are presented as mean±standard error of the mean (SEM). Each experiment was repeated twice or thrice with duplicate or triplicate data points each as indicated under the results section. Statistical analyses were conducted using SAS 9.1 (SAS Institute, Cary, NC). Statistical significance was determined by one way analysis of variance (ANOVA) followed by a Tukey test as a post-ANOVA multiple comparison test. Unpaired t-test was used to compare the two groups. P < 0.05 was considered statistically significant.
Results
Leptin increases DNA adducts formation in MCF-7 cells
Our results demonstrated that in MCF-7 cells, leptin significantly increased the depurinating DNA adducts 4-OHE1[E2]-1-N3 adenine and 4-OHE1 [E2]-1-N7 guanine levels compared with treatment with E2 alone. Thus, in MCF-7 cells treated with E2 alone or E2 plus leptin, the N3 adenine levels were 0.0025±0.0005 and 0.29±0.04 (pmol/106cells), respectively. Furthermore, the levels of 4-OHE1[E2]-1-N7 guanine were 0.0025±0.0005 and 1.05±0.043 in cells treated with E2 alone or E2 plus leptin, respectively (Figure 2).
Leptin affects estrogen metabolizing genes and their expression in MCF-7 cells
Leptin upregulates CYP1B1
Our study shows that the treatment of MCF-7 cells with leptin (10, 50, and 100 ng/mL) significantly increased CYP1B1 levels as indicated by the reporter assay by 68.8%, 87.8%, and 141.11%, respectively (Figure 3A). CYP1B1 protein was also upregulated as indicated by Western blot (Figure 3B).
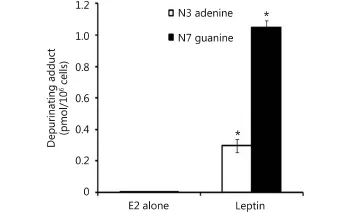
Figure 2 Effect of leptin on depurinating DNA adducts formation. Cells were grown in serum free media and treated with E2 (10 nM) alone or with leptin (5 ng/mL) for 48 hours. The media were collected for DNA adducts analysis by HPLC. * indicates significant difference of treated cells compared to control at P <0.05 using unpaired t-test.

Figure 3 Effect of leptin on CYP1B1 in MCF-7 cells. (A) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing CYP1B1 promoters. After treatment with different doses of leptin, luciferase production was analyzed 48 h later. (B) Western blot analysis. CYP1B1 protein levels were determined 72 h after treatment with different doses of leptin. * indicates significant difference of leptin treated cells compared to control at P < 0.05 using unpaired t-test. “a” indicates significant difference of cells treated with 100 ng/mL leptin compared with those treated with 10 ng/mL or 50 ng/mL using ANOVA.
Leptin downregulates NQO1
Our data reflected that the effect of leptin on NQO1 occurred on the transcriptional level as indicated by the Reporter assay and real time RT-PCR. In the Reporter assay, leptin (100 ng/mL) significantly downregulated NQO1 from 100% to 45.78% (Figure 4A). In the real time RT-PCR assay, NQO1 levels were reduced by 59.23% by using leptin100 ng/mL (Figure 4B). Furthermore, leptin decreased NQO1 on the translational level in MCF-7 cells as indicated by Western blot (Figure 4C).
Leptin downregulates COMT
Our Reporter gene assay demonstrated that leptin (10, 50, and 100 ng/mL) reduced COMTP1 expression by 20.45%, 20.32%, and 30.17%, respectively (Figure 5Ai). Similarly, COMTP2 was reduced by 37%, 31.76 %, and 37.08%, respectively (Figure 5Aii). Real time RT-PCR data reflected that COMT mRNA was reduced by 34.57% in cells treated with leptin (100 ng/mL) compared with untreated control (Figure 5B). Consistent with the above two assays, Western blot confirmed that leptin downregulated COMT expression compared with control (Figure 5C).

Figure 4 Effect of leptin on NQO1 in MCF-7 cells. (A) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing NQO1 promoters. After treatment with leptin (100 ng/mL), luciferase production was measured 48 h later. (B) Real time RT-PCR analysis. The cells were treated with leptin (100 ng/mL) for 24 h followed by total RNA isolation from the cells, and the NQO1 mRNA expression was determined by real time RT-PCR. The threshold cycle value of NQO1 was normalized based on that of GAPDH. (C) Western blot analysis. NQO1 protein levels were determined 72 h after treatment with different doses of leptin. * indicates significant difference of leptin treated cells compared to control at P < 0.05 using unpaired t-test.
Discussion
Although technological advances in medical sciences and health care have realized the detection and early treatment of breast cancer, several unanswered questions with regard to the mechanisms that underlie this disease persist.
Obese women are at risk of breast cancer; however, clinicians lack solid tools for the prevention or early diagnosis of this risk. Although the connection between obesity and breast cancer has been well documented in various studies10, the molecular mechanisms fundamental for this correlation are not fully clear. Leptin, an obesity related molecule, is a key player in the progression of several types of cancer including breast cancer. Leptin signaling increases cell proliferation in breast cancer via the induction of cyclin D1 and Cdk219,20and evading apoptosis21, as well as stimulation of angiogenesis22, epithelial mesenchymal transition (EMT)23, and telomerase activities24. Moreover, leptininduces Notch and IL-1 inflammatory systems25and influences several cell components of the breast cancer stroma (i.e., cancer associated fibroblasts, macrophages, adipocytes and cancer stem cells)16,26. Furthermore, tamoxifen was found to induce an increase in leptin expression which may be one of the causes of drug resistance among cancer patients treated with tamoxifen27.

Figure 5 Effect of leptin on COMT in MCF-7 cells. (A) Luciferase reporter gene assay. The cells were stably transfected with reporter plasmids containing COMTP1 (i) and COMTP2 promoters (ii). After treatment with different doses of leptin, luciferase production was analyzed 48 h later. Results represent the Mean±SEM of two experiments. (B) Real time RT-PCR analysis. The cells were treated with leptin (100 ng/mL) for 24 h followed by total RNA isolation from the cells and COMT mRNA expression was determined by real time RT-PCR. The threshold cycle value of COMT was normalized based on that of GAPDH. Results represent the Mean±SEM of 2 experiments. (C) Western blot analysis. Both S-COMT and MB-COMT protein levels were determined 72 h after treatment with different doses of leptin. * indicates significant difference of leptin treated cells compared to control at P < 0.05 using unpaired t-test.
From the above findings, it is clear that the research associated with obesity, leptin, and breast cancer progression becomes more complex with an overlap between several leptin mediated pathways. Increased studies are therefore necessary to fully elucidate the mechanisms by which leptin affects breast cancer. This study analyzed a novel potential mechanism by which leptin may influence breast tumor growth and development. Considering the crosstalk between leptin and estrogen, we focused on the metabolic pathways of estrogen, more specifically the estradiol metabolism and the several enzymes involved that have been linked to breast cancer. This study aims to inform possible future intervention to reduce the progression of breast cancer.
Our results demonstrated that leptin directs breast cancer cells toward the production of carcinogenic estrogen-DNA adducts N3-adenine and N7-guanine. To elucidate the mechanism by which this adduction occurs, the effect of leptin on the expression of some key enzymes involved in estrogen metabolism was examined. The expression levels of the CYP1B1, NQO1, and COMT enzymes were of special importance.
We found that leptin upregulated CYP1B1, which confirmed the effect of leptin on CYP1B1 as stated in previous studies. As previously mentioned, leptin induces the CYP1B1 expression in breast cancer cells. The mechanism of this induction through the activation of ERK and AKT lead to the ligand-independent activation of the ERα pathway28. Other studies have demonstrated the same effect of leptin on CYP1B1 in MCF-7 cells as well21. As mentioned, this effect is interesting because CYP1B1 is involved in the oxidative metabolism of estrogen (4-hydroxylation), an important step in the adduct formation pathway. We also found that leptin reduced the levels of the detoxifying enzyme COMT, an important protective enzyme against estrogen-induced carcinogenesis. This is in accordance with an interesting study that used proteomic analysis to study changes in whole MCF-7 cell proteome in response to leptin29. On the effect of leptin on NQO1, our study indicated that leptin reduced NQO1 levels. To our knowledge, no previous study has explored the relation between leptin and this enzyme. However, the high levels of NQO1 in human adipose tissueswhich indicated for the role of NQO1 in the metabolic complications of human obesity has been reported30. Thus, we determined that leptin upregulated CYP1B1 with a concomitant reduction of detoxifying enzymes COMT and NQO1. Clearly, this may have increased the levels of DNA adducts N3-adenine and N7-guanine. Interestingly, it was recently reported that leptin induces the proliferation of prostate cancer cells, and estrogen metabolism was identified as an essential co-player in this respect suggesting that leptin might be a novel target for the therapeutic intervention of prostatic disorders, which is pathophysiologically greatly similar to breast cancer31.
Several studies have been performed to elucidate on the role leptin plays on different breast cell lines both normal and cancerous. A study evaluated the influence of leptin on cell proliferation, cell cycle, apoptosis, and signaling on numerous breast cell lines, including 184B5 normal and MCF-10A fibrocystic cells, as well as MCF-7, MDA-MB-231, and T47D cancer cells. Leptin stimulated the proliferation of all cell lines except for 184B5 and MDA-MB-231 cells. In addition, leptin inhibited apoptosis but failed to alter the proportion of cells within the cell cycle in MCF-7 cells. Moreover, leptin induced the overexpression of leptin, Ob-R, estrogen receptor, and aromatase mRNA in MCF-7 and T47D cells. This finding suggests that obesity-associated hyperleptinemia may be a breast cancer risk factor32. Otvos et al.33used a peptide called Allo-aca, a drug targeting the leptin receptor (ObR), to see its effects on triple negative breast cancer (TNBC) xenograph model. Results were very promising and indicated exciting prospects on a possible therapeutic target for TNBC. However, minimal work has been conducted on the relationship between leptin and estrogen metabolic pathways as mentioned above, thereby confirming the novelty of the current work.
Collectively, these results extend the knowledge on estrogen/leptin crosstalk and implicate new potential mechanisms for obesity-associated breast cancer given that leptin is a major adipokine produced by adipocytes. This may pave the way for developing new agents that target leptin and leptin receptor pathways that shape the tumor microenvironment particularly in obese women and may encourage the personalized and professional control of obesity. Advances in adipokine research may hold promise for the use of adipokines as potential prognostic markers and therapeutic targets. Physical exercise, adoption of a balanced diet, weight reduction, and bariatric surgery for morbidly obese women may regulate adipokine levels to reduce the risks of developing breast cancer. Similarly, lipid-lowering drugs such as statins and niacin, vitamin C and D supplementation, folic acid, oleic acid, and calcium-channel blockers may also significantly modulate serum adipokines levels and would be of value in obesity associated breast cancer34.
This study collectively shows that leptin directs estrogen metabolism toward unfavorable pathways by increasing the formation of DNA adducts. These adducts can then generate mutations resulting in the progression of breast cancer. This finding may add a new avenue by which leptin may drive changes in breast cancer microenvironment. Moreover, the estrogen metabolic pathways may represent potential targets for the treatment and prevention of obesity-related breast cancer. Further studies should be directed toward elucidating the exact molecular mechanisms by which leptin alters estrogen metabolic pathways.
However, this study includes limitations including its focus on only breast cancer cells. Thus, it would be of great interest to investigate the effect of leptin on estradiol-induced adduct formation in normal epithelial cells (e.g., MCF-10A). The outcome of this study may provide information on whether the adducts formed will have a role in the risk of breast cancer. Future research can explore whether the estrogen metabolic pathway is involved in higher resistance to endocrine therapy among obese breast cancer patients who are resistant to endocrine therapy as well. Further evaluation of human breast cancer samples and experimental models, as well as the development of novel in vivo models designed to specifically study mammary carcinogenesis can certainly help address many of these questions.
Acknowledgements
This work was supported by a grant from University of Texas Medical Branch National Institute of Environmental Health Sciences Center Pilot Project E506676. We express our deep gratitude to Salama A. Salama (Department of Pharmacology, Alazhar University, Cairo, Egypt; previous instructor at University of Texas Medical Branch at Galveston) for the scientific advice and funds to complete this work. Moreover, we acknowledge the contribution of Barbara C. Spink (Wadsworth Center, New York State Department of Health, Albany, NY) who generously provided us with CYP1B1 Luciferase Reporter plasmids. In addition, we would like to thank Muhammad Saeed (Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, USA) for helping in the DNA adducts analysis.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Tao ZQ, Shi AM, Lu CT, Song T, Zhang ZG, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015; 72: 333-8.
2.Fussell KC, Udasin RG, Smith PJS, Gallo MA, Laskin JD. Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis. 2011; 32: 1285-93.
3.Ziegler RG, Faupel-Badger JM, Sue LY, Fuhrman BJ, Falk RT, Boyd-Morin J, et al. A new approach to measuring estrogen exposure and metabolism in epidemiologic studies. J Steroid Biochem Mol Biol. 2010; 121: 538-45.
4.Cavalieri EL, Rogan EG. Depurinating estrogen-DNA adducts, generators of cancer initiation: their minimization leads to cancer prevention. Clin Transl Med. 2016; 5: 12.
5.Zhao ZL, Kosinska W, Khmelnitsky M, Cavalieri EL, Rogan EG, Chakravarti D, et al. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat2 embryonic cells and the mutational spectrum of 4-hydroxyestradiol. Chem Res Toxicol. 2006; 19: 475-9.
6.Embrechts J, Lemière F, Van Dongen W, Esmans EL, Buytaert P, Van Marck E, et al. Detection of estrogen DNA-adducts in human breast tumor tissue and healthy tissue by combined nano LC-nano ES tandem mass spectrometry. J Am Sco Mass Spectrom. 2003; 14: 482-91.
7.Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011; 125: 169-80.
8.Cavalieri E, Rogan E. The molecular etiology and prevention of estrogen-initiated cancers: Ockham's Razor: Pluralitas non estponenda sine necessitate. Plurality should not be posited without necessity. Mol Aspects Med. 2014; 36: 1-55.
9.Kamel M, Shouman S, El-Merzebany M, Kilic G, Veenstra T, Saeed M, et al. Effect of tumour necrosis factor-alpha on estrogen metabolic pathways in breast cancer cells. J Cancer. 2012; 3: 310-21.
10.Reding KW, Zahid M, Cavalieri E, Rogan EG, Raccor BS, Atkinson C, et al. Associations between dietary intake of fruits and vegetables in relation to urinary estrogen DNA adduct ratio. Open J Prev Med. 2014; 4: 429-37.
11.Simone V, D'Avenia M, Argentiero A, Felici C, Rizzo FM, De Pergola G, et al. Obesity and breast cancer: molecular interconnections and potential clinical applications. Oncologist. 2016; 21: 404-17.
12.Vona-Davis L, Rose DP. Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 2009; 20: 193-201.
13.Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med. 2013; 64: 45-57.
14.Babaei Z, Moslemi D, Parsian H, Khafri S, Pouramir M, Mosapour A. Relationship of obesity with serum concentrations of leptin, CRP and IL-6 in breast cancer survivors. J Egypt Natl Canc Inst. 2015; 27: 223-9.
15.Blanquer-Rosselló MDM, Oliver J, Sastre-Serra J, Valle A, Roca P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int J Biochem Cell Biol. 2016; 72: 18-26.
16.Andò S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol. 2011; 8: 263-75.
17.Li WJ, Xu LZ, Chen Y, Mu L, Cheng M, Xu WM, et al. Effect of estrodiol on leptin receptors expression in regulating fat distribution and adipocyte genesis. Gynecol Endocrinol. 2016; 32: 464-8.
18.Saeed M, Rogan E, Fernandez SV, Sheriff F, Russo J, Cavalieri E. Formation of depurinating N3Adenine and N7Guanine adducts by MCF-10F cells cultured in the presence of 4-hydroxyestradiol. Int J Cancer. 2007; 120: 1821-4.
19.Gonzalez RR, Cherfils S, Escobar M, Yoo JH, Carino C, Styer AK, et al. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2). J Biol Chem. 2006; 281: 26320-8.
20.Saxena NK, Vertino PM, Anania FA, Sharma D. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J Biol Chem. 2007; 282: 13316-25.
21.Ray A, Nkhata KJ, Cleary MP. Effects of leptin on human breast cancer cell lines in relationship to estrogen receptor and HER2 status. Int J Oncol. 2007; 30(6): 1499-509.
22.Zhou W, Guo S, Gonzalez-Perez RR. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br J Cancer. 2011; 104: 128-37.
23.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires βcatenin activation via Akt/GSK3- and MTA1/Wnt1 proteindependent pathways. J Biol Chem. 2012; 287: 8598-612.
24.Ren H, Zhao TS, Wang XC, Gao CT, Wang J, Yu M, et al. Leptin upregulates telomerase activity and transcription of human telomerase reverse transcriptase in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2010; 394: 59-63.
25.Lipsey CC, Harbuzariu A, Daley-Brown D, Gonzalez-Perez RR. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J Methodol. 2016; 26; 6: 43-55.
26.Guo SC, Gonzalez-Perez RR. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One. 2011; 6: e21467.
27.Ozet A, Arpaci F, Yilmaz MI, Ayta H, Ozturk B, Komurcu S, et al. Effects of Tamoxifen on the serum leptin level in patients with breast cancer. Jpn J Clin Oncol. 2001; 31: 424-7.
28.Khanal T, Kim HG, Do MT, Choi JH, Won SS, Kang W, et al. Leptin induces CYP1B1 expression in MCF-7 cells through ligandindependent activation of the ERα pathway. Toxicol ApplPharmacol. 2014; 277: 39-48.
29.Valle A, Sastre-Serra J, Pol C, Miró AM, Oliver J, Roca P. Proteomic analysis of MCF-7 breast cancer cell line exposed to leptin. Anal Cell Pathol (Amst). 2011; 34: 147-57.
30.Palming J, Sjöholm K, Jernås M, Lystig TC, Gummesson A, Romeo S, et al. The expression of NAD(P)H: quinone oxidoreductase 1 is high in human adipose tissue, reduced by weight loss, and correlates with adiposity, insulin sensitivity, and markers of liver dysfunction. J Clin Endocrinol Metab. 2007; 92: 2346-52.
31.Habib CN, Al-Abd AM, Tolba MF, Khalifa AE, Khedr A, Mosli HA, et al. Leptin influences estrogen metabolism and accelerates prostate cell proliferation. Life Sci. 2015; 121: 10-5.
32.Dubois V, Jardé T, Delort L, Billard H, Bernard-Gallon D, Berger E, et al. Leptin induces a proliferative response in breast cancer cells but not in normal breast cells. Nutr Cancer. 2014; 66: 645-55.
33.Otvos L Jr, Kovalszky I, Riolfi M, Ferla R, Olah J, Sztodola A, et al. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur J Cancer. 2011; 47: 1578-84.
34.Dalamaga M. Obesity, insulin resistance, adipocytokines and breast cancer: New biomarkers and attractive therapeutic targets. World J Exp Med. 2013; 3: 34-42.
Cite this article as:Shouman S, Wagih M, Kamel M. Leptin influences estrogen metabolism and increases DNA adduct formation in breast cancer cells. Cancer Biol Med. 2016; 13: 505-13. doi: 10.20892/j.issn.2095-3941.2016.0079
Marwa Kamel
E-mail: marwawka@yahoo.com
Received September 20, 2016; accepted October 18, 2016.
Available at www.cancerbiomed.org
Copyright © 2016 by Cancer Biology & Medicine
 Cancer Biology & Medicine2016年4期
Cancer Biology & Medicine2016年4期
- Cancer Biology & Medicine的其它文章
- Liver cancer: challenge and prospect
- Role of microRNAs in inflammation-associated liver cancer
- Portal vein embolization for induction of selective hepatic hypertrophy prior to major hepatectomy: rationale, techniques, outcomes and future directions
- Three-dimensional printing: review of application in medicine and hepatic surgery
- Portal vein tumor thrombus is a bottleneck in the treatment of hepatocellular carcinoma
- Bcl-2 expression is a poor predictor for hepatocellular carcinoma prognosis of andropause-age patients