
草酸铜催化苯直接氧化制备苯酚
2017-01-12 01:05王伟涛石磊磊马养民任喜迎
陕西科技大学学报 2017年1期
王伟涛, 石磊磊, 马养民, 任喜迎
(陕西科技大学 化学与化工学院 教育部轻化工助剂化学与技术重点实验室, 陕西 西安 710021)
草酸铜催化苯直接氧化制备苯酚
王伟涛, 石磊磊, 马养民, 任喜迎
(陕西科技大学 化学与化工学院 教育部轻化工助剂化学与技术重点实验室, 陕西 西安 710021)
以H2C2O4和CuSO4通过沉淀法制备了草酸铜,并对所制备的化合物进行了X射线粉末衍射(XRD)、扫描电镜(SEM)和X射线光电子能谱(XPS)的表征.结果表明,制备草酸铜为斜方晶系的草酸铜.以制备的草酸铜为催化剂,研究了其对苯直接氧化制备苯酚的催化性能,并考察了催化剂量、H2O2量、反应时间等反应条件对该反应的影响.结果表明,在优化条件下,草酸铜催化剂可以催化双氧水氧化苯得到13.1%苯酚,并且该催化剂可以重复使用,回收率高.
草酸铜; 苯; 苯酚; 氧化
0 引言
苯酚是一种重要的化工原料,在有机合成、医药、农药、树脂等方面具有广泛的应用[1,2].目前苯酚的工业生产方法主要是异丙苯法,90%以上的苯酚是通过异丙苯法生产的[3].尽管如此,异丙苯法仍存在步骤多、工艺条件苛刻以及整体效率低等不足,且生产成本受副产物丙酮的价格而波动.随着绿色化学的发展,苯直接氧化制备苯酚以其方法简便、原子经济性高、环境友好等优点[4],引起了国内外的广泛研究[5-7].以H2O2作氧化剂,其反应副产物是水.因此,H2O2直接氧化苯制备苯酚被认为是最有希望取代异丙苯法制备苯酚的一种清洁生产工艺[8].
H2O2直接氧化苯制备苯酚的催化剂主要有杂多酸及其修饰的杂多酸类[9]、分子筛类[10]、过渡金属氧化物类[11-14]等.其中,过渡金属氧化物,以其优良的催化性能在苯直接氧化制备反应中得到广泛的研究[15-17],特别是铜氧化物的催化剂受到了高度的关注.Makgwane等[18]合成了CuFe氧化物催化剂,以H2O2为氧化剂,催化苯直接氧化制备苯酚的反应,苯的转化率达到44%,苯酚的选择性达到91%;但是催化剂的制备方法复杂,且制备温度较高.Yamada等[19]制备了[Cu(tmpa)]2+嫁接在Al-MCM-41的催化剂,并研究了其在室温下催化H2O2直接氧化苯合成苯酚的反应,得到的苯酚选择性近乎100%,但在其制备过程中合成配体较为困难.这些催化剂具有较好的催化效果,但是其制备方法复杂,催化剂稳定性差,重复使用性能差.因此,开发一种制备方法简单且稳定性好的催化剂对研究苯直接氧化制备苯酚反应具有重要的意义.
草酸铜具有稳定的化学性质,不易被氧化,几乎不溶于水、乙酸等溶剂.因此,以草酸铜为催化剂,以期望其在苯氧化制备苯酚反应中具有良好的催化性能和稳定性.此外,草酸铜是一种常见的铜盐,其合成方法简单,制备成本低,甚至废水中铜离子的回收就是利用草酸铜沉淀法[20].因此,本文通过沉淀法制备了草酸铜,将其用于苯直接氧化制备苯酚的反应,并考察了其催化苯直接氧化制备苯酚反应的影响因素及重复使用性能.
1 实验部分
1.1 试剂和仪器
(1)主要试剂:草酸,五水硫酸铜,H2O2(30%),分析纯,天津市天力化学有限公司;无水乙醇,分析纯,天津市河东区红岩试剂厂;苯,乙腈,甲醇,高效液相色谱纯,天津市科密欧化学试剂有限公司;实验中所用水均为去离子水.
(2)主要仪器:BS2202S电子天平(赛多利斯科学仪器北京有限公司);高速离心机(上海安亭科学仪器厂);DF-101S集热式恒温加热磁力搅拌器(郑州长城科工贸有限公司);高效液相色谱仪(WAYEELC 3000-2系列,MB-C18色谱柱,250 mm×4.60 mm).
1.2 草酸铜催化剂的制备
采用沉淀法制备草酸铜催化剂.具体过程如下:将一定量的五水硫酸铜加入若干水中,得到溶液A;将一定量的草酸加入若干水中,得到溶液B;然后将溶液B缓慢滴加到带有磁子搅拌的溶液A中,在室温下反应一段时间,将生成的沉淀抽滤,并用水和无水乙醇洗涤三次以上,在100 ℃干燥10 h,即可得到草酸铜催化剂.
1.3 催化剂表征和催化性能测试
(1)样品表征:采用Ultima IV X-射线粉末衍射仪对样品的晶相组成进行测定.电压40 kV,电流40 mA,功率1.6 kW,狭缝DS、RS和SS分别为1 mm、0.15 mm和1 °,扫描速度为8 °/min;采用Q 45环境扫描电子显微镜对样品的形貌进行测定,电压20 kV,电流15 mA;草酸铜中铜的结合能是采用多功能X射线光电子能谱仪(XPS)进行测定的.Al Kα激发源,靶电压和靶电流分别为15 kV和8 mA,真空室气压小于2×10-6Pa,分析器传输能量为50 eV,测量步长为0.1 eV,溅射速度为0.2 nm/s,溅射面积为 2 mm×2 mm.
(2)H2O2浓度测试:采用高锰酸钾直接滴定法测定H2O2的浓度.H2O2的分解率(C)按下式进行计算:

(3)催化性能测试:苯直接氧化制备苯酚反应的过程如下:将1.0 mL苯(11.25 mmol)和3.0 mL乙腈先加入到装有磁力搅拌子和回流冷凝管的50 mL烧瓶中,再加入100 mg催化剂,待升温至70 ℃时,逐滴加入5.0 mL H2O2,恒温搅拌反应4 h,反应结束后,转移至25 mL容量瓶中用甲醇定容.通过GC-MS确定反应产物,利用高效液相色谱进行分析,按下式计算苯酚产率:
2 结果与讨论
2.1 XRD分析
图1表示制备的草酸铜催化剂的XRD图谱.从图1可以发现,具有较强的衍射峰,2θ角分别为22.76 °、36.08 °、46.54 °、51.54 °,这些衍射峰与斜方晶系草酸铜晶体结构CuC2O4(JCPDS 21-0297)的衍射峰相吻合;这也与文献[21]制备的草酸铜的主要衍射峰基本一致.因此,可以确定合成的催化剂主要成分为斜方晶系草酸铜晶体.
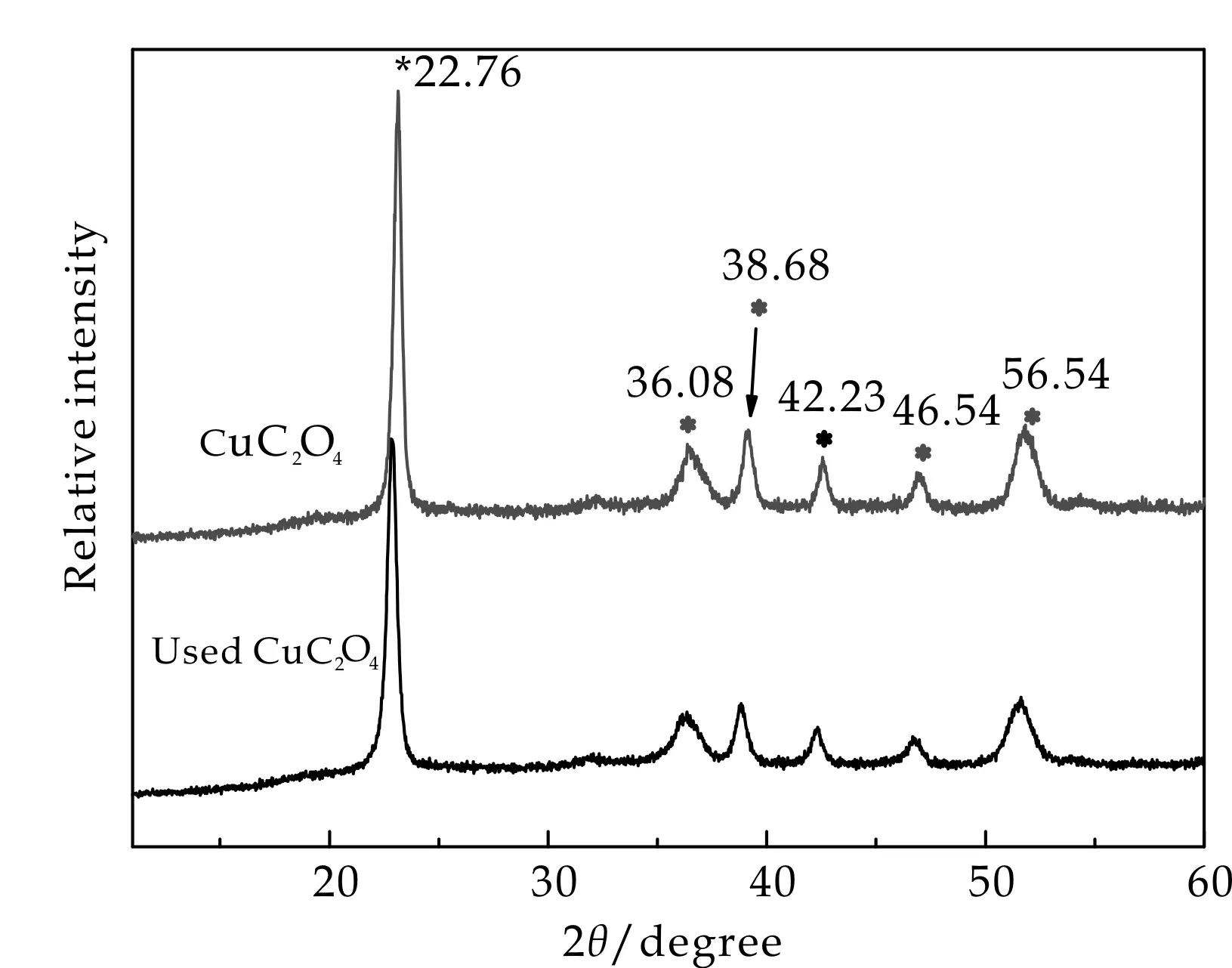
图1 新制备和使用过一次的草酸铜XRD图
2.2 SEM分析
图2为草酸铜粉末放大5 000倍的SEM照片.从图2(a)中可以发现,新制备的草酸铜粉末颗粒大小基本均匀,粒径比较小,而且存在一定程度的团聚现象.

(a)新制备的草酸铜SEM图

(b)使用过一次的草酸铜SEM图图2 草酸铜的SEM图
2.3 草酸铜催化性能研究
以草酸铜为催化剂,探讨反应条件对苯直接氧化反应的影响,并对重复使用性能进行了考察.
2.3.1 催化剂用量对苯直接氧化反应的影响
催化剂的用量影响着催化反应的性能.在保持其它条件不变,考察草酸铜催化剂用量对苯直接氧化制备苯酚反应的影响,实验结果如表1所示.从表1中可以看出,苯酚的产率随着催化剂用量的增加呈先增加后下降的趁势.当催化剂的量为100 mg时,苯酚的产率达到最大值为13.1%.这是由于过多的催化剂加快了H2O2的分解,产生过量的·OH,使苯酚进一步氧化,从而导致苯酚产率降低[22].为了证明这一点,考察了草酸铜催化剂对H2O2分解的影响,其结果如表2所示.从表2可以发现,在70 ℃,没有催化剂时,H2O2的分解率为16.8%;随着催化剂量的增加,H2O2的分解率升高.这表明草酸铜能够催化H2O2的分解.
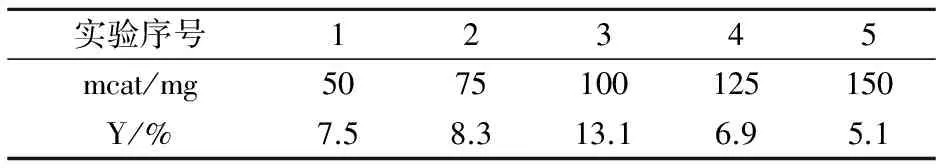
表1 催化剂量对苯直接氧化制备
反应条件: 1.0 mL苯, 3.0 mL乙腈,5.0 mL 30%的H2O2,70 ℃, 反应4 h.
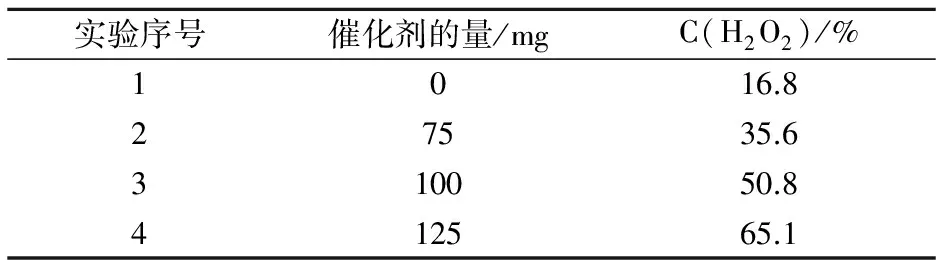
表2 催化剂对H2O2分解的影响实验
实验条件:3.0 mL乙腈,5.0 mL 30%的 H2O2,70 ℃,反应0.5 h.
2.3.2 H2O2用量对苯直接氧化反应的影响
H2O2作为氧化剂,是影响苯直接氧化反应的一个重要因素.改变H2O2的用量,草酸铜催化苯直接氧化反应的实验结果如表3所示.随着H2O2用量的增加,苯酚的产率明显提高;当H2O2用量超过5.0 mL(48.97 mmol)时,苯酚产率有所下降;这可能是由于过多的H2O2用量使苯酚被进一步氧化,从而导致苯酚的产率降低.当H2O2与苯的体积比为5∶1时,苯酚的产率为13.1%.理论上按化学计量比,1 mol 苯和1 mol H2O2恰好完全反应,但在实验中H2O2和苯的最优体积比为5∶1,对应的摩尔比为4.35∶1,远大于摩尔比1∶1,这是由于在反应过程中H2O2会不可避免的分解[23],使得反应中使用的H2O2的用量增多.

表3 H2O2用量对苯直接氧化制备
反应条件: 1.0 mL苯, 3.0 mL乙腈,100 mg 草酸铜,70 ℃, 反应4 h.
2.3.3 反应时间对苯直接氧化反应的影响
由于苯酚更容易被氧化,属于一个连续反应,因此,反应时间对苯直接氧化反应有重要的影响.在其它反应条件不变时,草酸铜催化苯直接氧化反应随时间的变化如表4所示.随着反应时间延长,苯酚的产率明显提高,进一步延长时间,苯酚的收率有所下降;这可能是过量的H2O2使苯酚进一步氧化生成副产物所致[12].从表4中可以看出,最优的反应时间为4 h.

表4 反应时间对苯直接氧化制备
反应条件:1.0 mL苯, 3.0 mL乙腈,100 mg草酸铜,5.0 mL 30%的H2O2,70 ℃.
2.3.4 溶剂对苯氧化反应的影响
溶剂是影响苯直接氧化制备苯酚的一个重要因素.由于乙酸作为溶剂比乙腈作溶剂的产率高[24],受到人们的关注.因此,考察了乙酸水溶液作为溶剂对反应的影响.以3.0 mL 70%(V/V)的乙酸水溶液为溶剂,其它条件不变的情况下,得到了不足1%的苯酚产率.这可能是由于乙酸提供了酸性环境及配位作用影响了苯氧化反应[22].
2.3.5 催化剂的重复使用
重复使用性能是考察催化剂的一个重要指标.在最优条件下考察了催化剂的重复使用性能.将使用过一次的催化剂洗涤、干燥后,直接用于下一次反应,在其他相同的条件下考察其催化性能.将催化剂重复使用5次,发现苯酚的产率分别为12.9%, 9.5%,8.9%,8.5%,8.3%,如表5所示.这表明催化剂能够循环使用,且回收率较高.尽管如此,循环使用后催化性能略有下降.这可能是由于一方面催化剂回收中存在不可避免的损失,另一方面是催化剂有所流失[25].
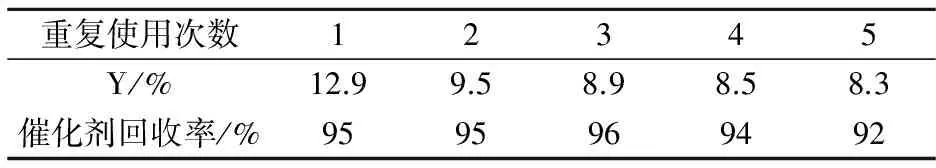
表5 催化剂的重复使用实验
反应条件: 1.0 mL苯, 3.0 mL乙腈,100 mg草酸铜,5.0 mL 30%的H2O2,70 ℃, 反应4 h.
通过XRD表征使用一次的催化剂,发现和新制备的催化剂衍射峰一致,均为斜方晶系草酸铜晶体(如图1所示);这说明使用一次后的催化剂仍然是草酸铜.通过SEM图(如图2(b)所示)发现使用一次的催化剂的粒径明显比未使用的催化剂的粒径小;这说明了草酸铜与H2O2发生了相互作用.在草酸铜催化H2O2的分解实验中,经观察实验现象发现,反应后的溶液为淡蓝色,而催化剂由淡蓝绿色变为黑色.这表明在H2O2下,草酸铜有微量流失到溶液中,同时草酸铜中有部分的一价铜产生.通过XPS对使用过一次的草酸铜进行表征,如图3所示.在结合能935 eV处的峰对应为Cu(Ⅱ),结合能在932.8 eV处的峰对应为Cu(I)[26],所以使用过一次的催化剂中存在Cu(I)和Cu(Ⅱ)[27].使用一次的催化剂中出现Cu(I)峰表明催化剂中的Cu2+有一部分变成了Cu+.所以,催化剂重复使用中催化性能的下降是由于H2O2和催化剂作用,Cu2+活性成分流失而造成的.
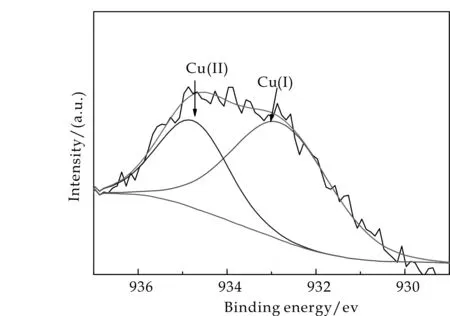
图3 使用一次草酸铜的Cu 2p XPS图
3 结论
采用沉淀法由铜盐和草酸制备草酸铜,该方法具有操作简单,成本低等优点.通过XRD、SEM、XPS等表征手段,证明了制备的化合物为斜方晶系草酸铜晶体,而且颗粒较小,粒径比较均匀.催化苯氧化实验表明,制备的草酸铜可用作苯直接氧化制备苯酚的催化剂,并且表现出较高的催化活性和催化剂稳定性.草酸铜催化苯直接氧化制备苯酚的最优反应条件为:苯(1.0 mL)、乙腈(3 .0mL)、草酸铜(100 mg)、H2O2(5 .0mL)、反应温度70 ℃、反应时间4 h,苯酚的产率为13.1%.因此,草酸铜可以作为一种成本低、稳定性好的催化剂催化苯直接氧化制备苯酚反应.
[1] 冯素姣,张 丽,任远航,等.Keggin型杂多酸铯盐催化苯氧化反应[J].化学学报,2012,70(22):2 316-2 322.
[2] 蒋斯扬,孔 岩,吴 丞,等.高Cu含量MCM-41在苯直接氧化反应中的催化性能[J].催化学报,2006,27(5):421-426.
[3] 王 晓,张天永,姜 爽,等.含铁催化剂催化苯直接氧化制备苯酚的研究进展[J].化工进展,2015,34(2):381-388.
[4] 翟庆辉,刘 欢,顾婷婷,等.Keggin结构磷钼钒酸催化苯直接氧化[J].聊城大学学报(自然科学版),2014,27(2):52-55.
[5] Xu D,Liu L L,Zhao Z L,et al.Preparation and characterization of mesoporous Ag/VO(x)-TiO2employed for catalytic hydroxylation of benzene[J].Journal of Nanoscience & Nanotechnology,2014,14(6):242-246.
[6] Liu H,Lu G Z,Guo Y L,et al.Effect of pretreatment on properties of TS-1/diatomite catalyst for hydroxylation of phenol by H2O2in fixed-bed reactor[J].Catalysis Today,2004,93-95(3):353-357.
[7] Wang D K,Wang M T,Li Z H.Fe-based metal-organic frameworks for highly selective photocatalytic benzene hydroxylation to phenol[J].Acs Catalysis,2015,5(11):6 852-6 857.
[8] 任永利,米镇涛.过氧化氢氧化苯制苯酚的催化剂研究进展[J].化工进展,2002,21(11):827-830.
[9] Yuan C Y,Gao X H,Pan Z S,et al.Molybdovanadophosphoric anion ionic liquid as a reusable catalyst for solvent-free benzene oxidation to phenol by H2O2[J].Catalysis Communications, 2015, 58:215-218.
[10] Guo B,Zhu L,Hu X,et al.Nature of vanadium species on vanadium silicalite-1 zeolite and their stability in hydroxylation reaction of benzene to phenol[J].Catalysis Science & Technology,2011,1(6):1 060-1 067.
[11] Guo B Y,Bo S,Yan P,et al.FexOy@C spheres as an excellent catalyst for Fischer-Tropsch synthesis[J].Journal of the American Chemical Society,2010,132(3):935-937.
[12] Borah P. VOPO4·2H2O encapsulated in graphene oxide as a heterogeneous catalyst for selective hydroxylation of benzene to phenol[J].Green Chemistry,2016,18(2):397-401.
[13] Ma Y M,Ren X Y,Wang W T,et al.Hydroxylation of benzene to phenol on CuxOy@C with hydrogen peroxide[J].Reaction Kinetics Mechanisms & Catalysis,2016,117(2):693-704.
[14] Wang W T,Ding G D,Jiang T,et al.Facile one-pot synthesis of VxOy/C catalysts using sucrose for the direct hydroxylation of benzene to phenol[J].Green Chemistry,2013,15(5):1 150-1 154.
[15] Hu L Y,Yue B,Chen X Y,et al.Direct hydroxylation of benzene to phenol on Cu-V bimetal modified HMS catalysts[J].Catalysis Communications,2014,43(2):179-183.
[16] Xu D,Jia L H,Guo X F.Cu-doped mesoporous VO-TiO2in catalytic hydroxylation of benzene to phenol[J].Chinese Journal of Catalysis,2013,34(2):341-350.
[17] Samia A K,Islam H E M,Lateefa A,et al.Preparation and surface characterization of CuO and Fe2O3catalyst[J].Applied Surface Science,2012,258(19):7 617-7 624.
[18] Makgwane P R,Ray S S.Hydroxylation of benzene to phenol over magnetic recyclable nanostructured CuFe mixed-oxide catalyst[J].Journal of Molecular Catalysis A Chemical,2015,398:149-157.
[19] Yamada M,Karlin K D,Fukuzumi S.One-step selective hydroxylation of benzene to phenol with hydrogen peroxide catalysed by copper complexes incorporated into mesoporous silica-alumina[J].Chemical Science,2016,7(4):2 856-2 863.
[20] 杨 焰,李德良,邓瑞帅.微蚀废液中铜回收工艺研究[J].中南林业科技大学学报(自然科学版),2010,30(5):160-162.
[21] 石 磊,李金英,赵志军,等.反萃沉淀法制备草酸铜超细粉体[J]. 原子能科学技术,2010,44(11):1 292-1 298.
[22] Acharyya S S.Facile synthesis of CuCr2O4spinel nanoparticles:A recyclable heterogeneous catalyst for the one pot hydroxylation of benzene[J].Catalysis Science & Technology,2014,4(12):4 232-4 241.
[23] Tang Y,Zhang J.Direct oxidation of benzene to phenol catalyzed by vanadium substituted heteropolymolybdic acid[J].Transition Metal Chemistry,2006,31(3):299-305.
[24] Chen J,Gao S,Xu J.Direct hydroxylation of benzene to phenol over a new vanadium-substituted phosphomolybdate as a solid catalyst[J].Catalysis Communications,2008,9(5):728-733.
[25] Tanarungsun G,Kiatkittipong W,Praserthdam P,et al.Ternary metal oxide catalysts for selective oxidation of benzene to phenol[J].Journal of Industrial & Engineering Chemistry,2008,14(5):596-601
[26] Ghodselahi T,Vesaghi M A,Shafiekhani A,et al.XPS study of the Cu@Cu2O core-shell nanoparticles[J].Applied Surface Science,2008,255(5):2 730-2 734.
[27] Martínez J M L,Rodríguez Castelln E,Snchez R M T,et al.XPS studies on the Cu(I,II)-polyampholyte heterogeneous catalyst:An insight into its structure and mechanism[J].Journal of Molecular Catalysis A Chemical,2011,339(1-2):43-51.
【责任编辑:陈 佳】
Direct hydroxylation of benzene to phenol on copper oxalate
WANG Wei-tao, SHI Lei-lei, MA Yang-min, REN Xi-ying
(College of Chemistry and Chemical Engineering, Key Laboratory of Auxiliary Chemistry & Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science & Technology, Xi′an 710021, China)
Copper oxalate was prepared by the precipitation method with H2C2O4and CuSO4.The prepared copper oxalate was characterized by XRD,SEM and XPS.It revealed that prepared compound was the copper oxalate and was in the orthorhombic system.Its catalytic performance was investigated by the reaction of direct hydroxylation of benzene to phenol.The reaction conditions,which included the dosage of catalyst,the amount of H2O2,the reaction time, were optimized. Under the optimal condition, the yield of phenol was 13.1%.Furthermore, the catalyst could be reused with the high recovery.
copper oxalate; benzene; phenol; hydroxylation
国家自然科学基金项目(21403136); 陕西省科技厅自然科学基础研究计划项目(2016JQ2025); 陕西科技大学博士科研启动基金项目(BJ13-26)
王伟涛(1985-),男,陕西扶风人,讲师,博士,研究方向:绿色催化
1000-5811(2017)01-0097-05
TQ243.1
A
猜你喜欢
环境卫生工程(2021年5期)2021-11-20
云南化工(2020年11期)2021-01-14
合成技术及应用(2021年1期)2021-01-07
材料科学与工程学报(2016年4期)2017-01-15
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
当代化工研究(2016年2期)2016-03-20
合成化学(2015年4期)2016-01-17
天津城建大学学报(2015年5期)2015-12-09
化工进展(2015年6期)2015-11-13
