
表面增强拉曼光谱法检测人尿液、粪便中马来酸依那普利的浓度及其临床应用Δ
2016-12-19 08:24:18赖青鸟吴超权广西壮族自治区食品药品检验所南宁5300华北理工大学基础医学院药理教研室唐山053009
中国药房 2016年32期
甘 盛,赖青鸟,韩 婷,吴超权(.广西壮族自治区食品药品检验所,南宁 5300;.华北理工大学基础医学院药理教研室,唐山 053009)
表面增强拉曼光谱法检测人尿液、粪便中马来酸依那普利的浓度及其临床应用Δ
甘盛1*,赖青鸟1,韩婷2,吴超权1(1.广西壮族自治区食品药品检验所,南宁530021;2.华北理工大学基础医学院药理教研室,唐山053009)
目的:建立检测人尿液和粪便中马来酸依那普利浓度的方法,并将其应用于临床。方法:尿液和粪便样品经前处理后,与表面信号增强剂胶体银溶胶按1∶1(V/V)的比例混合,采用表面增强拉曼光谱法检测。激发功率为400 mW,激发波长为785 nm,分辨率为4.5 cm-1,用于定性的拉曼位移为(800±2)、(1 050±2)、(1 330±2)、(1 450±2)和(1 630±2)cm-1,用于定量的拉曼位移为(1 050±2)cm-1。结果:马来酸依那普利尿药、粪药浓度分别在0.50~100.00 μg/ml和2.00~100.00 μg/g范围内线性关系良好(r分别为0.989 3和0.978 0,n=6),定量下限分别为0.50 μg/ml和2.00 μg/g,检测限分别为0.10 μg/ml和1.30 μg/g;日内、日间RSD<5%,方法回收率分别为95.24%~99.38%、86.73%~94.57%,提取回收率分别为84.29%~90.43%(RSD=0.84%)、82.36%~87.70%(RSD=1.84%)。采用该法检测15例高血压患者马来酸依那普利的尿药浓度为15.31~33.46 μg/ml,粪药浓度为5.08~41.79 μg/g;疗效判定为“有效”的患者平均尿药、粪药浓度与“未见好转”的患者比较,差异均无统计学意义(P>0.05)。结论:表面增强拉曼光谱法用于人尿液和粪便中马来酸依那普利浓度的检测简便、快速。
马来酸依那普利;尿液;粪便;表面增强剂;表面增强拉曼光谱法
依那普利为强效血管紧张素Ⅰ转换酶抑制剂,其抑制作用是卡托普利的8倍,临床上一般应用其马来酸盐,其适用于各种程度的高血压、肾血管性高血压和糖尿病合并高血压患者的临床治疗(单独或与其他药物联合使用)[1],尤其适用于常规应用洋地黄或利尿药难于控制者,并可延缓充血性心力衰竭的临床进展[2]。目前,对该药体内消除情况的监测一般采用高效液相(HPLC)法或液质联用(LC-MS)法,但这类方法操作烦琐、成本较高,且需采集患者血样。拉曼光谱法具有操作简便、测定时间短、无需采血等优点[3-5],但由于其灵敏度低、检测限难以达到体内微量分析的要求,故在实际应用中需加入拉曼信号表面增强剂(一般为粒径较大的等金属离子溶胶)吸附待测物分子,以增强其拉曼散射信号[4-5]。相对于传统的血药浓度监测方法,表面增强拉曼光谱法仅以患者的排泄物作为检测对象,大大简化了样品的前处理过程,减轻了患者的痛苦。因此,本试验以人尿液和粪便样品作为研究对象,采用表面增强拉曼光谱法检测其中马来酸依那普利的质量浓度,探索体内药物浓度测定的新方法,为临床治疗药物监测和合理用药提供参考。
1 材料
1.1仪器
OPAL-3000型便携式拉曼光谱仪,包括HRP-8型高光通量光线探头、热电制冷低温电荷耦合阵列检测器(CCD,可在-60℃下稳定工作,检测灵敏度为15~50 ppm)(英国Metage公司);HLA-1024型超声波清洗机(深圳华龙超声机电设备有限公司);Vortex-Genie 2型涡旋振荡器(美国Scientific Industries公司);R201BV型旋转蒸发仪(上海申胜生物技术有限公司);X85-2型可控温磁力搅拌器(上海梅颖浦仪器仪表制造有限公司);TDL-5-A型离心机(上海安亭科学仪器厂)。
1.2药品与试剂
马来酸依那普利对照品(中国食品药品检定研究院,批号:100705-201203,纯度:98.87%);马来酸依那普利片(上海现代制药股份有限公司,批号:20140953,规格:5 mg);玻璃纤维滤膜、聚四氟乙烯滤膜(湖北津华量化环境检测科技有限公司);WAX固相萃取柱(美国Phenomenex公司);甲醇为色谱纯,水为去离子水,其余试剂为分析纯。空白尿液和粪便由华北理工大学附属医院提供。
2 方法与结果
取马来酸依那普利对照品约10 mg,精密称定,置10 ml量瓶中,加水5 ml,涡旋振荡5 min,超声(功率为1 200 W)10 min,加水至刻度,摇匀,配制成相当于马来酸依那普利质量浓度为1 mg/ml的对照品贮备液,密封、避光保存。取上述对照品贮备液适量,用水稀释得相应质量浓度的标准溶液,备用。
2.2尿液和粪便样品的处理
2.2.1尿液样品取新鲜尿液200 ml,以离心半径6.75 cm、转速5 000 r/min离心5 min,取上清液5 ml,加入硼酸盐缓冲液(pH=10.3)0.5 ml,混匀,加入乙酸乙酯15 ml,涡旋混合1 min,以离心半径6.75 cm、转速5 000 r/min离心10 min,收集上清液于圆底烧瓶中,用乙酸乙酯提取2次,合并提取液,于42℃下旋转蒸干,残渣用50%甲醇1 ml复溶,即得尿液样品的待测溶液,备用。
2.2.2粪便样品将新鲜粪便冷冻干燥24 h后,研磨均匀,去除较大的粗纤维。精密称取1.0 g置于50 ml具塞玻璃离心管中,加入甲醇20 ml,超声(功率为1 200 W)提取10 min,重复提取3次,合并提取液,于4℃下以离心半径13.5 cm、转速10 000 r/min离心20 min,取上清液于45℃下旋转蒸发,将其浓缩至1 ml,经0.45 μm微孔滤膜滤过后,滤液用水稀释至50 ml。将上述溶液上样至已活化的WAX固相萃取柱中,用丙酮-0.1 mol/L碳酸氢钠溶液(40∶60,V/V)6 ml以5 ml/min的速度洗脱,待无液体流出后抽真空5 min;再用乙酸乙酯8 ml以1 ml/min的速度洗脱,合并2次洗脱液,常温下以氮气流吹扫,将其浓缩至1 ml。将此浓缩液转移至硅胶柱中,用丙酮-甲醇(1∶1,V/V)以1 ml/min的速度洗脱,洗脱液于常温下以氮气流吹干,残渣用50%甲醇1 ml复溶,即得粪便样品的待测溶液,备用。
2.3表面增强剂和上机液的制备
应用柠檬酸钠还原法[7]合成胶体银溶胶。取水99 ml置于三口圆底烧瓶中,加入1%硝酸银水溶液1 ml,加热至微沸(92~93℃),快速加入1%柠檬酸三钠水溶液4 ml,冷凝回流,采用可控温磁力搅拌器使之保持沸腾。于95℃时停止加热,冷却并保持均匀搅拌,静置25 min,得粒径为30~40 nm的胶体银溶胶。取上述胶体银溶胶和“2.2.1”“2.2.2”项下的待测溶液各0.5 ml,分别涡旋混匀,即得尿液和粪便样品的上机液。
2.4检测条件
2.4.1定性检测采用拉曼光谱仪对上述上机液进行扫描。激发功率:400 mW;激发波长:785 nm;分辨率:4.5 cm-l;马来酸依那普利特征峰的定性拉曼位移:(800±2)、(1 050±2)、(1 330±2)、(1 450±2)和(1 630±2)cm-l。
模块是模块化设计的基本元素,是一种实体的概念,如把模块定义为一组同时具有相同功能和相同结合要素,具有不同性能或用途甚至不同结构特征,但能互换的单元[2-5].模块化一般是指使用模块的概念对设备或系统进行规划设计、生产组织.设备的模块化设计是在对一定范围内的不同功能或相同功能不同性能、不同规格的设备进行功能结构分析的基础上,划分并设计出一系列模块,通过模块的选择和组合可以构成不同的设备,以满足发射场不同需求的设计方法.
2.4.2定量检测[8]以(1 050±2)cm-l对应峰强度进行谱图归一化。以(1 050±2)cm-l对应峰强度与已知贮备液对应峰强度相比,计算马来酸依那普利的质量浓度。c/c贮=Iv/I贮(式中,c为待测物质量浓度,c贮为贮备液质量浓度,Iv为待测物特征峰强度,I贮为贮备液特征峰强度)。
2.5方法学考察
2.5.1标准曲线的制备取空白尿液和对照品贮备液各适量,分别配制成马来酸依那普利质量浓度为0.50、1.00、5.00、10.00、25.00、50.00、100.00 μg/ml的尿液样品,按“2.2.1”“2.3”项下方法处理后,进样分析,记录拉曼光谱图,见图1B、1C。以拉曼光谱强度(I尿)为纵坐标、待测物质量浓度(c尿)为横坐标进行线性回归,得回归方程为I尿=6 265.24c尿-217.70(r尿=0.989 3,n=6)。结果表明,马来酸依那普利的尿药浓度在0.50~100.00 μg/ml范围内线性关系良好,其定量下限为0.50 μg/ml,检测限为0.10 μg/ml。
取空白粪便和对照品贮备液各适量,分别配制成马来酸依那普利质量浓度为2.00、5.00、10.00、25.00、50.00、100.00 μg/g的粪便样品,按“2.2.2”“2.3”项下方法处理后,进样分析,记录拉曼光谱图,见图1D、1E。以拉曼光谱强度(I粪)为纵坐标、待测物质量浓度(c粪)为横坐标进行线性回归,得回归方程为I粪=4 872.31c粪+659.01(r粪=0.978 0,n=6)。结果表明,马来酸依那普利的粪药浓度在2.00~100.00 μg/g范围内线性关系良好,其定量下限为2.00 μg/g,检测限为1.30 μg/g。
2.5.2精密度与准确度试验取空白尿液、空白粪便和对照品贮备液各适量,分别配制成马来酸依那普利低、中、高质量浓度(1.00、10.00和50.00 μg/ml)的尿液样品和低、中、高质量浓度(5.00、10.00和50.00 μg/g)的粪便样品,分别按“2.2”和“2.3”项下方法处理后,进样测定。每质量浓度取9样本分析,连续测定7 d,根据当日标准曲线计算各样品的测得质量浓度,考察方法的精密度与准确度。结果显示,各尿液样品的日内、日间RSD<3%,方法回收率为95.24%~99.38%;各粪便样品的日内、日间RSD<5%,方法回收率为86.73%~94.57%。精密度与准确度试验结果见表1。
表1 精密度与准确度试验结果(±s)Tab 1 Results of precision and accuracy tests(±s)
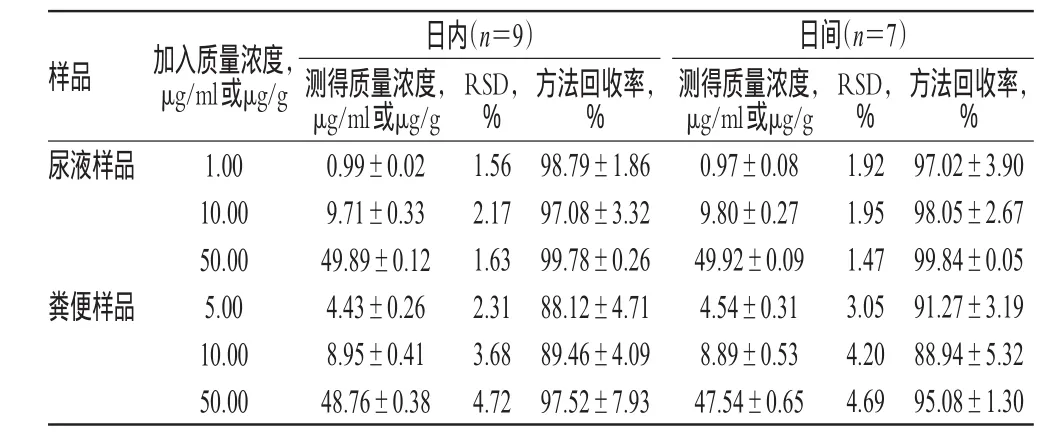
表1 精密度与准确度试验结果(±s)Tab 1 Results of precision and accuracy tests(±s)
样品 加入质量浓度,μg/ml或μg/g尿液样品粪便样品1.00 10.00 50.00 5.00 10.00 50.00日内(n=9)测得质量浓度,μg/ml或μg/g 0.99±0.02 9.71±0.33 49.89±0.12 4.43±0.26 8.95±0.41 48.76±0.38 RSD,% 1.56 2.17 1.63 2.31 3.68 4.72方法回收率,% 98.79±1.86 97.08±3.32 99.78±0.26 88.12±4.71 89.46±4.09 97.52±7.93日间(n=7)测得质量浓度,μg/ml或μg/g 0.97±0.08 9.80±0.27 49.92±0.09 4.54±0.31 8.89±0.53 47.54±0.65 RSD,% 1.92 1.95 1.47 3.05 4.20 4.69方法回收率,% 97.02±3.90 98.05±2.67 99.84±0.05 91.27±3.19 88.94±5.32 95.08±1.30
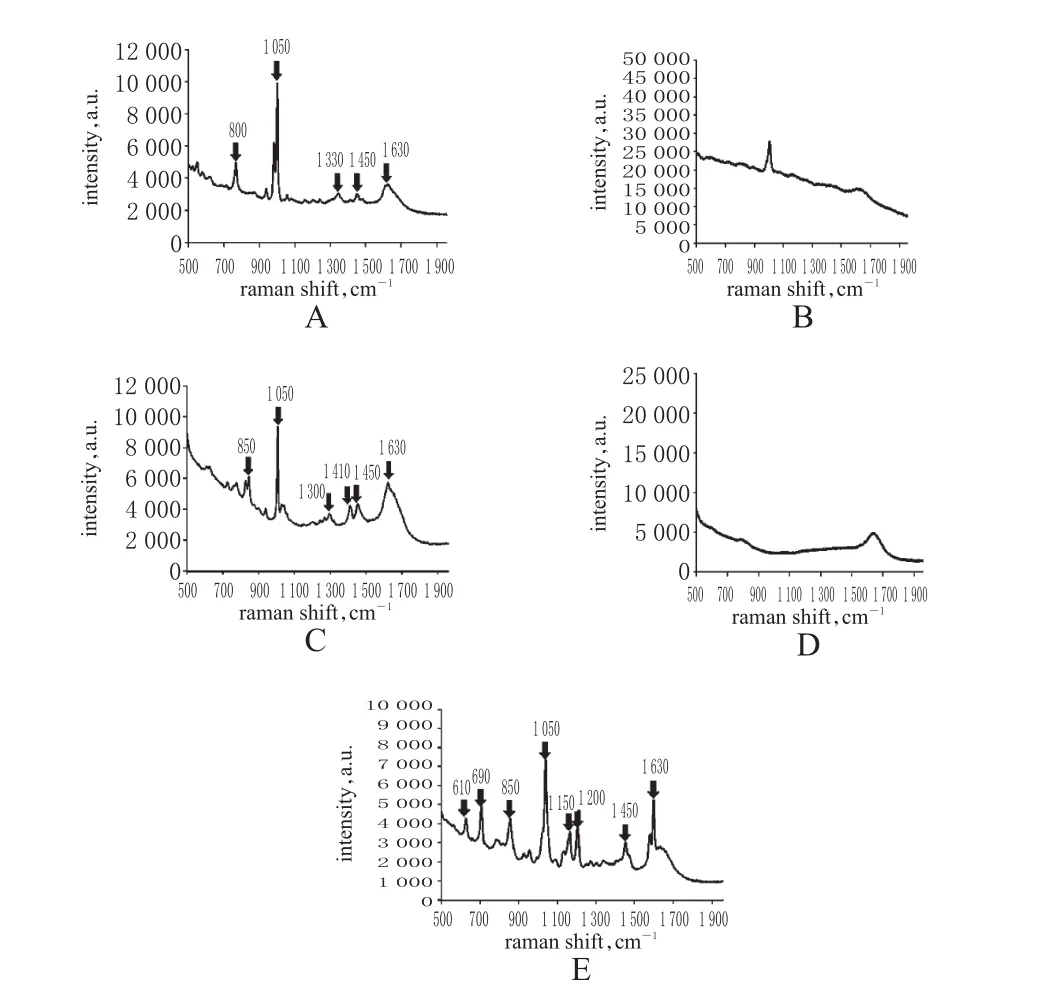
图1 马来酸依那普利的拉曼光谱图A.标准溶液(10.00 μg/ml);B.空白尿液;C.空白尿液+马来酸依那普利;D.空白粪便;E.空白粪便+马来酸依那普利Fig 1 Raman spectrum of enalapril maleateA.standard solution(10.00 μg/ml);B.blank urine;C.blank urine+enalapril maleate;D.blank faeces;E.blank faeces+enalapril maleate
2.5.3提取回收率试验取空白尿液、空白粪便和对照品贮备液各适量,分别配制成马来酸依那普利低、中、高质量浓度(1.00、10.00和50.00 μg/ml)的尿液样品和低、中、高质量浓度(5.00、10.00和50.00 μg/g)的粪便样品,分别按“2.2”和“2.3”项下方法处理后,进样测定;另取空白尿液和空白粪便各适量,分别按“2.2”和“2.3”项下方法处理后,加入对照品贮备液适量,使最终质量浓度与前者对应,进样测定。每质量浓度取6样本分析,考察方法的提取回收率。结果显示,尿液样品的提取回收率为84.29%~90.43%(RSD=0.84%),粪便样品的提取回收率为82.36%~87.70%(RSD=1.84%)。
2.5.4稳定性试验取空白尿液、空白粪便和对照品贮备液各适量,分别配制成马来酸依那普利低、高质量浓度(1.00和50.00 μg/ml)的尿液样品和低、高质量浓度(5.00和50.00 μg/g)的粪便样品,分别于0、2、4、6、9、12、15和24 h进样测定,考察方法的稳定性。结果显示,各样品在24 h内稳定,RSD<5%。
2.6临床应用
2.6.1样品来源选择华北理工大学附属医院初入院的高血压患者15例,年龄43.1~67.8岁,体质量46.9~71.5 kg,血尿常规、心电图、肝功能正常,胃肠功能良好。入院前3天及住院期间未服用过任何血管紧张素Ⅰ转换酶抑制剂,并禁烟、酒、茶[9]。本研究方案经华北理工大学附属医院医学伦理委员会审批,所有患者均知情同意并签署知情同意书。
2.6.2样品测定和统计学方法所有患者每日口服马来酸依那普利片20 mg,每日2次,分别于用药的第1~7天收集其尿液和粪便,分别按“2.2.1”“2.2.2”项下方法处理后,将所得待测溶液与表面增强剂胶体银溶胶按1∶1(V/V)的比例均匀混合采用表面增强拉曼光谱法检测其尿液和粪便中马来酸依那普利的质量浓度。采用SPSS 13.0软件处理所有数据。计量资料以x ±s表示,组间比较采用t检验。P<0.05为差异有统计学意义。
2.6.3样品测定结果检测结果显示,患者尿药浓度为15.31~33.46 μg/ml,粪药浓度为5.08~41.79 μg/g。大部分患者服药3 d后,尿药和粪药浓度不再大幅上升或下降,此时尿药和粪药浓度已趋于稳定。15例高血压患者马来酸依那普利尿药、粪药浓度的检测结果见表2。
表2 15例高血压患者马来酸依那普利尿药、粪药浓度的检测结果(±s ,n=15)Tab 2 Urinal and fecal concentrations of enalapril maleatein 15hypertensive patients(±s, n=15)
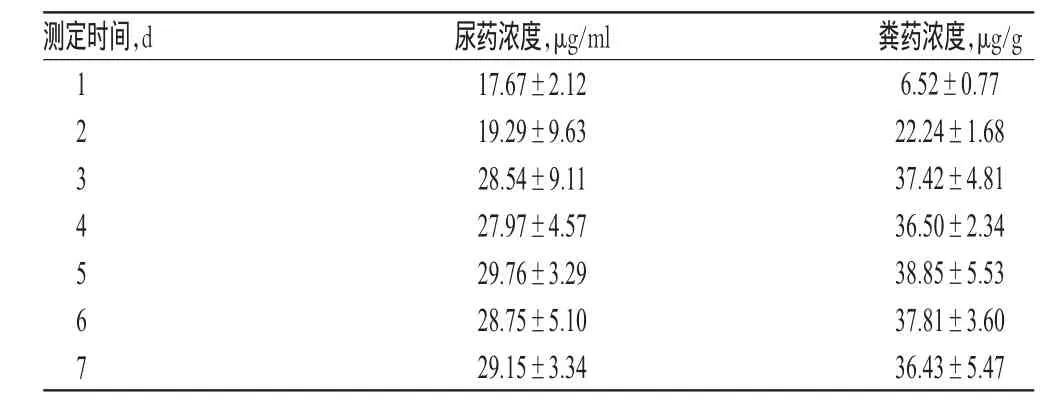
表2 15例高血压患者马来酸依那普利尿药、粪药浓度的检测结果(±s ,n=15)Tab 2 Urinal and fecal concentrations of enalapril maleatein 15hypertensive patients(±s, n=15)
测定时间,d 1234567尿药浓度,μg/ml 17.67±2.12 19.29±9.63 28.54±9.11 27.97±4.57 29.76±3.29 28.75±5.10 29.15±3.34粪药浓度,μg/g 6.52±0.77 22.24±1.68 37.42±4.81 36.50±2.34 38.85±5.53 37.81±3.60 36.43±5.47
以收缩压≤140 mm Hg(1 mm Hg=0.133 kPa)、舒张压≤90 mm Hg为标准(非同日连续3次测量)[10],对患者的治疗效果进行评价。15例患者入院3 d后,11例患者的血压符合上述标准,判定为“有效”;4例患者的血压仍较高,判定为“未见好转”。疗效判定为“有效”的患者平均尿药浓度为(28.14± 4.76)μg/ml,“未见好转”的患者平均尿药浓度为(26.77±5.91)μg/ml,组间比较差异无统计学意义(P>0.05);疗效判定为“有效”的患者平均粪药浓度为(37.65±4.20)μg/g,“未见好转”的患者平均粪药浓度为(35.09±5.65)μg/g,组间比较差异无统计学意义(P>0.05)。
3 讨论
3.1定量拉曼位移的筛选
在拉曼光谱图上,血管紧张素Ⅰ转换酶抑制剂的分子特征峰位于1 448~1 452 cm-1[苯环(C=C)伸缩振动]和1 627~1 633 cm-1[乙氧基(CH3-CH2-O-)弯曲振动]处。但由于各药物分子吡咯烷上的取代基不同,其特征峰的拉曼位移有所差异。马来酸依那普利的特征峰位于(800±2)cm-1和(1 330± 2)cm-1处,分别为吡咯烷上C-N伸缩振动和N-H伸缩振动。虽然尿素分子中的键谱峰覆盖了依那普利在相同拉曼位移处由C-OH非对称伸缩引起的振动峰,但对马来酸依那普利在(1 050±2)cm-1处特征峰的辨认不会造成大的干扰。随着待测物质量浓度的升高,此处峰强不断增加。因此,将定量拉曼位移确定为(1 050±2)cm-1,并以此处对应峰强度进行谱图归一化,计算尿液和粪便样品中马来酸依那普利的质量浓度。结果显示,马来酸依那普利的尿药和粪药浓度分别在0.50~100.00 μg/ml和2.00~100.00 μg/g范围内线性关系良好(r分别为0.989 3和0.978 0)。
3.2增强剂的选择[7]
表面增强剂的种类、质量浓度和混合比例均可影响拉曼光谱的信号增强效果及其检测限。笔者曾尝试使用金溶胶作为拉曼光谱信号增强剂,但并无明显的信号增强效果,在尿液与粪便样品中无法检测到待测物。而银溶胶则可明显增强待测物的信号。金属离子的空间密度与吸附分子的能力直接相关,过密则对吸附目标形成阻挡,过于分散则大量待测分子无法得到吸附。在可见光区域内,金的表面增强因子比银小,银溶胶与目标分子结合后可产生更为强烈的共振现象,即银溶胶与金溶胶相比,具有更强的表面增强活性。通过多次预试验,最终确定银溶胶与待测物溶液按1∶1等体积混合的效果最佳。
3.3银溶胶的稳定性
在试验过程发现,银溶胶在-4℃下保存3周后会出现沉淀。银溶胶的稳定性可能受以下因素影响:(1)在制备金属溶胶的过程中,使用了柠檬酸三钠,柠檬酸根离子不仅可作为还原剂,同时还具备稳定剂的效果,可提高金属溶胶的稳定性。柠檬酸三钠有3个羧基基团,其中2个吸附在银粒子的表面,余下的1个则通过其所带负电荷相互排斥的作用力起着稳定的作用。(2)温度对银溶胶的稳定性也有重要的影响。在室温下,纳米颗粒间的相互碰撞几率高于低温环境,易发生聚集、沉淀现象,不利于胶体的分散。(3)时间也是影响银溶胶稳定性的重要因素之一。长时间放置会使溶胶的稳定性下降,产生自凝聚现象。银溶胶在室温条件下,5~10 d内即会沉降失活。因此,银溶胶不宜长时间保存,建议低温保存,且配制后应尽快使用。
3.4尿液、粪便样品的检测限
马来酸依那普利在尿液样品中的检测限为0.50 μg/ml,粪便样品中的检测限为1.30 μg/g。尿液样品的检测限主要受其中尿素C-N π键对吡咯烷特征峰的影响;而粪便样品的检测限和线性关系则与其中杂质较多、影响待测物的辨认有关。
3.5与色谱法的比较[11]
与本方法相比,传统的色谱法耗时较长,单个样品从前处理到最后得出结果,色谱法至少需要1 h,而光谱法只需20 min左右。如果是对大批量样品进行检测,拉曼光谱法单个样品的扫描时间仅需几十秒,相对于色谱法而言,大大地节省了检测时间,因此能达到快速分析、检测的目的。在方法检测限方面,虽然较色谱法尚有一定的差距,但由于排泄物中的药物含量往往大大高于检测限,故仍可满足临床监测的需求。在方法准确度上,色谱法属于半确证性方法,通常需要与质谱串联以进一步确证结构。而拉曼光谱属于振动光谱,提供的是待测物分子的“指纹”信息,具有较高的准确性,但须在仪器的激发光强度、信噪比处理、检测器灵敏度上进行提高,同时对前处理进行优化,使检测方法更加完善[12]。
综上所述,与已有HPLC、气质联用(GC-MS)法和液相色谱-串联质谱(LC-MS/MS)法等比较[11,13-14],本研究所建立的表面增强拉曼光谱法,以患者的尿液和粪便样品为监测对象,大大简化了样品前处理过程,缩短了检测时间,降低了检测成本,并提高了患者的诊疗舒适度,为体内药物浓度的监测提供了新的方法。
[1]张金花,魏平.马来酸依那普利联合阿替洛尔治疗慢性心力衰竭的临床观察[J].中国药房,2015,26(3):314.
[2]吴伟.依那普利治疗慢性心衰的效果观察[J].数理医药学杂志,2016,29(1):78.
[3]陶琴,董健,钱卫平.表面增强拉曼光谱在定量分析中的应用[J].化学进展,2013,25(6):1 031.
[4]刘锋,朱勇,李铮.基于拉曼光谱评估人类精子存活能力的研究[J].生殖与避孕,2016,36(2):156.
[5]甘盛,赖青鸟,李志成,等.人尿液中盐酸氯丙那林的表面增强拉曼光谱法测定[J].中国医药工业杂志,2016,47(4):464.
[6]杨天鸣,黄国贞,付海燕,等.复杂生物样品中马来酸依那普利的快速分析新方法[J].化学与生物工程,2015,32(9):65.
[7]翟福丽,黄轶群,王锡昌,等.应用表面增强拉曼光谱技术快速检测尿样中的β-兴奋剂[J].分析化学,2012,40(5):718.
[8]吴雷,李菲,金周雨,等.基于密度泛函理论方法的核酸碱基拉曼光谱研究[J].生物化学与生物物理进展,2016,43(3):281.
[9]Lee C,Chun J,Hwang SW,et al.Enalapril inhibits nuclear factor-κB signaling in intestinal epithelial cells and peritoneal macrophages and attenuates experimental colitis in mice[J].Life Sci,2014,95(1):29.
[10]莫艾.马来酸依那普利叶酸片对于轻、中度高血压患者降压、降HCY的临床效果与安全性[J].中国实用医药,2016,11(3):141.
[11] 张勇,施振国.LC-MS/MS法同时测定人血浆中依那普利和依那普利拉的浓度[J].实用药物与临床,2015,18(12):1 469.
[12]Vodinh T,Hiromoto MYK,Begun GM,et al.Surface-enhanced raman spectrometry for trace organic analysis[J]. Anal Chem,2002,56(9):1 667.
[13]Shioya H,Shimojo M,Kawahara Y.Determination of enalapril and its active metabolite enalaprilat in plasma and urine by gaschromatography/mass spectrometry[J].Biomed Chromatogr,1992,6(2):59.
[14]王星,王琳,张廷剑,等.HPLC法同时测定马来酸依那普利与尼群地平的含量[J].实用药物与临床,2015,18(12):1 478.
(编辑:张元媛)
Concentration Determination of Enalapril Maleate in Human Urine and Feces by Surface-enhanced Raman Scattering Spectrometry and Its ClinicalApplication
GAN Sheng1,LAI Qingniao1,HAN Ting2,WU Chaoquan1(1.Guangxi Zhuang Autonomous Region Institute for Food and Drug Control,Nanning 530021,China;2.Pharmacology Teaching and Research Section,Preclinical College,North China University of Science and Technology,Hebei Tangshan 053009,China)
OBJECTIVE:To establish the method for concentration determination of enalapril maleate in human urine and feces,and apply it in the clinic.METHODS:The urine and feces samples were collected and pre-processed,then tested by surfaceenhanced Raman scattering spectrometry with ratio of sample to surface signal enhancer silver sol 1∶1(V/V).The assay was carried out with the following parameters:excitation power 400 mW,excitation wavelength 785 nm,resolution 4.5 cm-1.The characteristic shift of enalapril maleate were(800±2),(1 050±2),(1 330±2),(1 450±2),(1 630±2)cm-1.The quantitation shift was(1 050±2)cm-1.RESULTS:The linear range of enalapril maleate were 0.50-100.00 μg/ml in urine and 2.00-100.00 μg/g in feces(r=0.989 3,0.978 0,n=6);the limits of quantitation were 0.50 μg/ml and 2.00 μg/g,and the limits of detection were 0.10 μg/ ml and 1.30 μg/g.RSDs of intra-day and inter-day were both lower than 5%.The method recoveries were 95.24%-99.38%and 86.73%-94.57%;the extraction recoveries were 84.29%-90.43%(RSD=0.84%)and 82.36%-87.70%(RSD=1.84%),respectively.The urinal concentration of enalapril maleate in 15 hypertensive patients ranged 15.31-33.46 μg/ml,while the fecal concentration ranged 5.08-41.79 μg/g.There was no statistical significance in average urinal concentration and fecal concentration between“effective”patients and“no response”patients.CONCLUSIONS:Surface enhanced Raman scttering spectrometry can be used for the concentration determination of enalapril maleate in human urine and feces.It is simple and rapid.
Enalapril maleate;Urine;Feces;Surface signal enhancer;Surface-enhanced Raman scattering spectrometry
R917
A
1001-0408(2016)32-4504-04
10.6039/j.issn.1001-0408.2016.32.13
广西自然科学青年基金资助项目(No.2014GXNSF BA118059);中国食品药品检定研究院中青年发展研究基金项目(No.2013WC3)
*副主任药师,博士,硕士生导师。研究方向:食品药品检验及质量安全。电话:0771-5827256。E-mail:gansheng@hotmail.com
(2015-12-04
2016-06-16)
猜你喜欢
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
电子测试(2018年18期)2018-11-14 02:30:36
中国医药指南(2017年3期)2017-11-13 02:55:58
光学精密工程(2016年1期)2016-11-07 09:01:00
中国当代医药(2015年36期)2015-03-11 20:03:28
中国药业(2014年19期)2014-05-17 03:12:22
卫生职业教育(2014年16期)2014-05-16 03:48:28
济源职业技术学院学报(2014年3期)2014-02-28 02:35:42
河南医学研究(2014年3期)2014-02-27 14:51:45
中国现代应用药学(2013年4期)2013-03-11 19:13:53
