
构音障碍起病的1型强直性肌营养不良症1例报告并文献复习
2016-12-19 07:58李春荣吴秀娟房绍宽刘亢丁
中风与神经疾病杂志 2016年6期
李春荣, 吴秀娟, 房绍宽, 刘亢丁
构音障碍起病的1型强直性肌营养不良症1例报告并文献复习
李春荣, 吴秀娟, 房绍宽, 刘亢丁
强直性肌营养不良症(dystrophia myotonia,DM)是一种慢性、进展性常染色体显性遗传性疾病[1],可累及多系统如骨骼肌、晶状体、肺脏、心脏、胃肠道、中枢和周围神经系统、内分泌系统等,其临床表现呈多样化,通常以骨骼肌强直、无力和萎缩为主要表现。现将1例基因确诊的以构音障碍起病的伴有颅内多发白质病变的非遗传性DM1报道如下。
1 病例报告
患者,男,28岁,进行性言语不清6 y,双手无力2 y入院。患者于6 y前无明显诱因出现进行性言语不清,曾多次于当地医院就诊,行耳鼻喉内窥镜检查,未见明显异常,应用中药治疗(具体不详)后病情未见明显好转。头部MRI(2014-12-8):双侧额顶叶多发异常信号,考虑为脱髓鞘病变(见图1)。既往史:入伍8 y,诉正常参加训练。4 y前出现间断腹泻,诊断为慢性肠炎。家族史:父母体健,其母亲的妹妹2岁时仍不能行走,颈软,当地医院诊断为软骨病,6岁时去世。入院查体:血压:125/82 mmHg,心率92次/分,面窄、发稀,神清,构音障碍,双眼外展不到位;双侧额纹消失,双眼睑闭合无力,双侧鼓腮无力;伸舌居中,舌肌萎缩;左大鱼际肌萎缩,双手用力握拳后放松困难,四肢肌张力降低,四肢腱反射未引出,余神经系统查体未见异常。双侧胸锁乳突肌萎缩,双乳腺增大。辅助检查:性激素6项测定:孕酮(PROG) 0.30 nmol/L(正常值:0.138~2.671 nmol/L),促黄体激素(LH) 9.518 mIU/ml(正常值:1.24~8.62 mIU/ml),维生素B12:107.59(正常值:133~675 pmol/L);肌酶、血常规、凝血常规、肝、肾功能、垂体5项、超敏C反应蛋白、24 h游离皮质醇、糖化血红蛋白均未见异常。心电图:提示I度房室传导阻滞,ST段抬高(Ⅱ、Ⅲ、avf)。肌电图:上下肢可见肌强直放电。肌肉病理(右肱二头肌):可见肌纤维大小不等,萎缩肌纤维呈小角形,I型肌纤维为主,可见坏死及再生肌纤维,部分肌纤维肌膜核增加,可见部分核内移,未见肌浆块,肌间结缔组织无明显增加,未见炎性细胞浸润。NADH、SDH染色可见小径I型肌纤维,部分肌纤维酶活性缺失。GT染色未见RRF。COX、Acid、PAS、ORO未见明显异常(见图2、图3)。基因检测:送检血样本,基因 P12片段可见单个等位基因峰,基因P134片段可见三碱基梯度片段峰,超出正常范围。三核苷酸CTG重复次数分别为13和>600次(DM1正常重复次数为CTG≤50次)(见图4),符合DM1致病特征。阴囊彩超、心脏彩超、乳腺彩超未见明显异常。眼底检查未见异常。结合病史、体征及相关辅助检查临床诊断:强直性肌营养不良症1型,给予静点肌氨肽苷、七叶皂甙钠、舒血宁、肌注腺苷钴胺、口服艾地苯醌、B族维生素、叶酸片治疗,患者住院治疗4 d后出院,出院后口服B族维生素、艾地苯醌、叶酸片,5 m后随访患者从事保安一职,可正常参加日常跑步等体育训练。
2 讨 论
依据其致病基因的不同,DM分为DM1型和DM2两型,其中DM1型以肢体远端肌肉受累为主,其致病基因位于19q13区域DMPK基因3’端非翻译区CTG异常扩增引起;DM2型比DM1型少见,是以肢体近端肌肉受累为主,其致病基因位于3q21区域ZNF9基因内含子中的CCTG异常扩增引起[2]。DM1型是最常见的遗传性神经肌肉疾病[3], 其病因及发病机制未完全清楚,其致病基因位于19q13区域DMPK基因3’端非翻译区CTG异常扩增引起,针对异常扩增的CTG重复序列导致DM1多系统功能障碍机制的研究,主要有3种模型,目前主流的致病模型为异常扩增的CTG转录成有毒性的GUC RNA重复序列[4],折叠成发卡结构,滞留在细胞核内,形成核内聚集体,从而破坏基因的正常表达和可变剪接过程,这些异常导致许多肌肉和神经蛋白表达发生改变[5,6],从而引起多系统功能异常。
图1 头部MRI:提示双侧额叶、顶叶皮髓质交界区多发异常信号T2高信号(如黑箭所示)
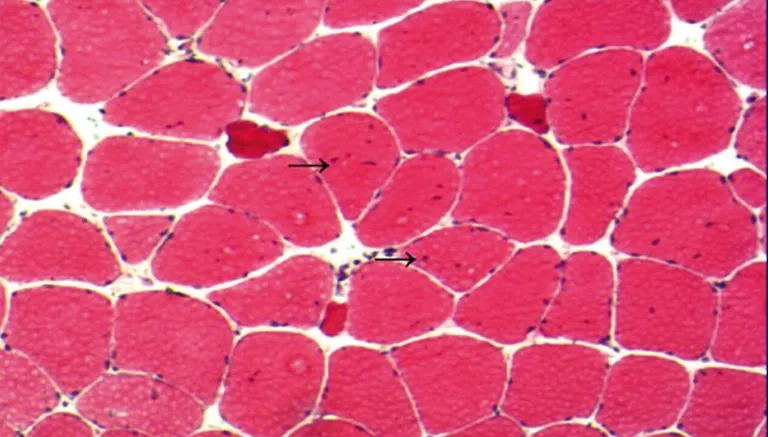
图2 肌肉病理:HE染色可见肌纤维大小不等,萎缩肌纤维呈小角形,I型肌纤维为主,可见坏死及再生肌纤维,部分肌纤维肌膜核增加可见部分核内移(如黑箭所示),未见肌浆块,肌间结缔组织无明显增加,未见炎性细胞浸润
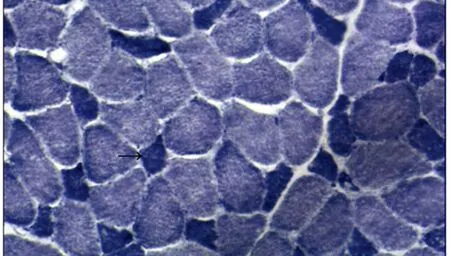
图3 NADH染色可见小径I型肌纤维(如黑箭所示),部分肌纤维酶活性缺失
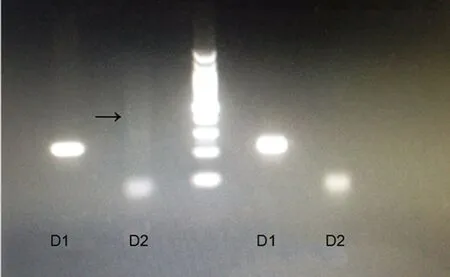
图4 左D1、D2为该患者基因图,右D1、D2为正常对照组的基因图
D1代表:常规PCR方法,扩增人两条染色体中,正常的一个;D2代表:采用TP-PCR方法,扩增人两条染色体中,异常的一个,对于患者因为TP-PCR引物设计在CTG重复区域,可以扩增一系列条带如图中黑箭所示区域
DM主要的临床表现为骨骼肌强直、无力和萎缩,其中肌强直最为常见,肌强直最常见双手握拳后不能立即松开,需重复数次才能放松,亦可出现呼吸肌和吞咽肌肌强直而导致通气不足,肌强直通常在肌萎缩之前数年或同时发生;肌萎缩往往先累及手部和前臂肌肉,继而累及头面部肌肉,如提上睑肌、颞肌、咬肌、面部诸肌、胸锁乳突肌等[7],咀嚼肌及面肌萎缩可表现为斧形脸,咬肌的萎缩可导致颞下颌关节脱位、张口受限等。另外DM患者常合并多系统损害,累及心肌者常出现心律失常、传导阻滞,严重的心电图改变如快速型心律失常可成为DM1型成年患者猝死的高危因素[8],累及消化系统可出现胃肠蠕动减慢,直肠蠕动异常可造成肠痉挛性腹痛,而胆囊排空延长导致胆结石[5];内分泌系统受累可出现脱发、糖耐量异常、生殖器发育不良、男性女性化、女性月经不规律等[9];累及中枢神经系统者可出现智力低下、嗜睡,头部可见颅骨内板增厚、蝶鞍变小、脑萎缩、局部或弥漫性脑白质损害[10];此外DM患者还可出现白内障并通常伴有视网膜色素变性。DM通常需与先天性肌强直鉴别。先天性肌强直有两种遗传类型:Thomsen 病-常染色体显性遗传,Becker病-常染色体隐性遗传[11]。DM与先天性肌强直的鉴别点主要包括先天性肌强直通常伴有肌肥大,而肌无力和肌萎缩不明显,且不会出现骨骼肌以外系统受累的表现,先天性肌强直致病基因为CLCN1位于染色体7q35。
目前DM的诊断主要依据临床表现、阳性家族史、肌电图、肌肉活检结果及基因检查结果。本例患者以慢性进行性言语不清为主要表现,数年后逐渐出现肌强直表现,病程中不伴有明显的肢体无力及萎缩,亦无家族遗传史,这是导致其多次就诊于不同医院但仍诊断不明确的主要原因。该患者在我们诊断DM1之前行头部MRI提示双侧额、顶叶皮髓交界区多发点片状异常信号(见图1),曾给我们定位诊断增加了困难,到目前为止,我国报道的DM1患者出现白质受损的病例很少,其病因和发病机制仍然不明确[12]。认知障碍的程度与白质受损的关系目前亦存在争议[13]。该患者并没有嗜睡、智力低下、情绪低落等临床表现,因此对于DM1型患者即使临床无神经系统受累的症状,条件允许情况下应该进一步完善头部MRI检查。DM1型患者的神经心理状态也应引起临床医生的重视。该患者同时伴有多系统损害的表现,如头发稀少、双侧乳腺增大为内分泌系统受累的表现;患者既往长期腹泻病史考虑可能由于DM累及消化道平滑肌引起胃肠蠕动减慢,胃肠排空慢引起;另外此患者还伴有循环系统(房室传导阻滞)及经头部MRI证实的中枢神经系统受累的表现。该患者临床表现不典型,肌电图结果虽提示肌强直放电,但是肌肉活检的结果未发现特征性病理表现,另外无阳性家族史,为其诊断增加了困难,最终基因检查的结果帮助确诊。
在DM的诊断中,由于临床表现复杂多样,单纯依靠临床表现很难做出诊断,虽然阳性家族史、肌肉活检具有一定的提示意义,但仍有其局限性,基因检查对DM确诊具有重要的作用。
该病目前尚无特殊治疗方法,苯妥英钠、卡马西平等膜稳定剂能促进钠泵活动,降低细胞膜内钠离子浓度,提高静息电位,改善肌强直症状;物理治疗对保持肌肉功能有一定作用;注意心脏病的监测和处理,白内障可手术治疗,内分泌异常可予相应处理;基因治疗是今后研究的方向。DM预后取决于发病年龄,幼年发病者预后较差,多在未成年死亡,成年发病者预后较好,可不影响寿命。
[1]Nigro G,Papa AA,Politano L.The heart and cardiac pacing in Steinert disease[J].Acta Myol,2012,31(2):110-116.
[2]Finsterer J,Rudnik-Schneborn S.Myotonic dystrophies:clinical presentation,pathogenesis,diagnostics and therapy[J].Fortschr Neurol Psychiatr,2015,83(1):9-17.
[3]Wissocque L,Brigadeau F,Richardson M,et al.Impairment of global and regional longitudinal strains in patients with myotonic dystrophy type 1[J].Int J Cardiol,2015,191:46-47.
[4]Gladman JT,Mandal M,Srinivasan V,et al.Age of onset of RNA toxicity influences phenotypic severity:evidence from an inducible mouse model of myotonic dystrophy (DM1)[J].PLoS One,2013,8(9):e72907.
[5] Lee JE,Bennett CF,Cooper TA.RNase H-mediated dcgradation of toxic RNA in myotonic dystrophy type 1[J].Proc Natl Acad Sci USA,2012,109(11):4221-4226.
[6] Meola G,Cardani R.Myotonic dystrophies:An update on clinical aspects,genetic,pathology,and molecular pathomechanisms [J].Biochim Biophys Acta,2015,52(4):594-606.
[7]Udd B,KraheR.The myotonic dystrophies:molecular,clinical,and therapeutic challenges[J].Lancet Neurol,2012,11(10):891-905.
[8]Groh WJ,Groh MR,Saha C,et al.Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1[J].N Engl J Med,2008,358(25):2688-2697.
[9]Meola G,Sansone V.Cerebral involvement in myotonic dystrophies[J].Muscle Nerve,2007,36(3):294-306.
[10] Kornblum C,ReulJ,KressW,et al.Cranial magnetic resonance imaging in genetically proven myotonic dystrophy type 1 and 2[J].Neurol,2004,251(6):710-714.
[11] Portaro S,Altamura C,Licata N,et al.Clinical,Molecular,and functional characterization of CLCN1 mutations in three families with recessive myotonia congenita[J].Neuromolecular Med,2015,17(3):285-296.
[12]Liu L,Liu HM,Liu ZJ,et al.Myotonic dystrophy type 1 associated with white matter hyperintense lesions:clinic,imaging,and genetic analysis[J].Chin Med J (Engl),2015,128(10):1412-1414.
[13] Minnerop M,Weber B,Schoene-Bake JC,et al.The brain in myotonic dystrophy 1 and 2:evidence for a predominant white matter disease[J].Brain,2011,134(12):3530-3546.
1003-2754(2016)06-0565-02
R746.2
2016-04-11;
2016-05-30
(吉林大学白求恩第一医院神经内科和神经科学中心,吉林 长春 130021)
刘亢丁,E-mail:kangdingliu@163.com;房绍宽,E-mail:fang20063536@sina.com
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
保健与生活(2022年13期)2022-07-06
疯狂英语·新读写(2021年10期)2021-12-07
国际放射医学核医学杂志(2021年10期)2021-02-28
奥秘(2019年8期)2019-08-28
武警医学(2019年2期)2019-03-05
食品安全导刊(2018年36期)2018-05-25
商周刊(2017年7期)2017-08-22
小猕猴智力画刊(2016年6期)2016-05-14
