
致病疫霉组蛋白H2A变异体序列与表达
2016-12-12 05:02汪晓雯国立耘
生物工程学报 2016年11期
汪晓雯,国立耘
中国农业大学 植物保护学院植物病理系 农业部植物病理学重点实验室,北京 100193
农业生物技术
致病疫霉组蛋白H2A变异体序列与表达
汪晓雯,国立耘
中国农业大学 植物保护学院植物病理系 农业部植物病理学重点实验室,北京 100193
汪晓雯, 国立耘. 致病疫霉组蛋白H2A变异体序列与表达. 生物工程学报, 2016, 32(11): 1564-1575.
Wang XW, Guo LY. Sequence analysis and expression patterns of histone H2A variants in Phytophthora infestans. Chin J Biotech, 2016, 32(11): 1564-1575.
在真核生物中,DNA缠绕在组蛋白上形成核小体,一个组蛋白分子包括H2A、H2B、H3和H4各2个核心组蛋白亚基。在这4种核心组蛋白中,H2A富含多样化,且在细胞的生物途径中起重要作用的变异体,因此,H2A家族一直是研究热点。致病疫霉是重要的病原菌也是研究卵菌的模式物种之一,目前关于卵菌表观遗传的研究还未见报道。本研究针对致病疫霉组蛋白H2A变异体,利用基因组信息和基因芯片数据,通过序列比对、系统发育分析以及基因表达水平检测,发现在致病疫霉基因组中存在组蛋白H2A变异体H2A.X.1、H2A.X.2和H2A.Z,它们在不同生长发育阶段和侵染过程呈现特异的表达谱。研究结果为进一步研究致病疫霉表观遗传机制奠定了基础。
卵菌,晚疫病菌,H2A.X,H2A.Z,系统发育,表达谱
在真核生物细胞中,组蛋白分子由H2A、H2B、H3和H4四个亚基组成,2个组蛋白分子组成1个八聚体的组蛋白,每147 bp DNA缠绕在1个组蛋白八聚体上形成核小体,通过连接组蛋白H1进而组装成染色质[1]。组蛋白的成分和它的共价修饰决定了核小体的状态,进而决定了染色质的状态。常规组蛋白 (Canonical histones) 在基因复制 (Gene duplication) 的过程中发生了个别位点的变异,进而演化为具有特异功能的组蛋白变异体。这些变异体在序列、理化性质、以及与核小体结合能力等方面与常规组蛋白有差异。因此,组蛋白的成分除了常规的H2A、H2B、H3和H4,还包括组蛋白变异体 (Histone variants)。真核生物的表观遗传机制包括组蛋白水平和DNA水平的修饰,组蛋白变异体及组蛋白的修饰共同参与维持染色质的结构和调控基因表达等过程。目前发现,组蛋白H2A和H3的变异体较多,并且广泛存在于各种真核生物细胞中[2]。组蛋白H2A的变体有H2A.X、H2A.Z、MacroH2A、H2A.Bbd等[3]。不同的H2A变异体差异主要在于C端尾巴的长度和序列,C端序列的变化决定了H2A变异体的功能[3]。
研究发现,H2A.X的功能是参与DNA损伤修复和维持基因组稳定。当DNA出现断裂时,H2A.X发生磷酸化修饰,替代常规H2A,进而将断裂的DNA双链进行非同源末端链接(Non-homologous end joining)[4]。H2A.X的磷酸化位点在酵母中是129位的丝氨酸残基,在哺乳动物中是139位的丝氨酸残基[2]。另外,H2A.X的高磷酸化也是细胞凋亡的特征之一[5]。同时,研究发现没有DNA双链断裂时也存在磷酸化的H2A.X,推论染色质的变化也能导致H2A.X的磷酸化,H2A.X参与稳定染色质结构[6]。
H2A.Z广泛分布于真核生物的染色体中,主要参与调节基因的转录激活,稳定核小体的结构,防止基因沉默蔓延到相邻的常染色质上[7]。研究发现H2A.Z通常结合在基因的启动子和转录起始位点 (TSSs)[8]。H2A.Z-H2B二聚体与DNA的结合相对较松,对转录激活起到重要的作用[9]。缺失H2A.Z,组蛋白去乙酰化酶Sir2、Sir3和Sir4将进入常染色质,基因转录活性降低[10]。
MacroH2A和H2A-Bbd仅在脊椎动物和哺乳动物体内发现。MacroH2A大量富集在失活的X染色体中,其C端含有很长的尾巴,形成球状结构域,抑制转录因子结合[2,11]。H2A-Bbd与组蛋白H4乙酰化同时出现,标志着染色体的转录激活,而在失活的X染色体中缺乏H2A-Bbd[2]。
疫霉Phytophthora属于卵菌 (Oomycetes),它和金褐藻、硅藻一并归属于茸鞭生物界(Stramenopila)[12]。疫霉是一类能生存于土壤,靠侵染寄主植物进行寄生生活的植物病原菌。致病疫霉Phytophthora infestans de Bary可侵染多种茄属植物,其中包括马铃薯和番茄[13]。致病疫霉引起的马铃薯晚疫病是马铃薯生产中的重要病害,曾经导致著名的爱尔兰饥馑。目前,该病害每年仍在世界范围内发生并造成严重危害。
尽管卵菌中多个物种的基因组数据已释放,但目前还缺少对组蛋白变异体的研究。疫霉菌的表观遗传机制也不清楚。依据已释放的基因组数据,本实验室在前期研究中发现,在卵菌基因组中含有保守的组蛋白H2A、H2B、H3和H4,并且H2A和H3存在序列保守的变异体。本研究利用基因组信息,对致病疫霉组蛋白H2A变异体进行序列、系统发育以及表达水平的分析,为进一步探索致病疫霉组蛋白H2A的功能建立基础。
1 材料与方法
1.1 材料
本试验中选取的供试菌株分别为2007年采自甘肃的致病疫霉A1交配型菌株MX5-1和YZ-6[14],以及来自美国的A2交配型菌株80787-94L[15]。培养致病疫霉菌株的培养基为黑麦番茄固体培养基和碗豆汁液体培养基[16]。RNA提取试剂盒NucleoSpin RNA Plant Kit购自MACHEREY-NAGEL公司。反转录酶和实时荧光定量PCR试剂均购自TaKaRa公司。引物由北京华大基因科技股份有限公司合成。聚碳酸酯膜购自Millipore公司。
1.2 方法
1.2.1 数据库搜索和基因序列分析
从NCBI、SWISS-PROT和KEGG数据库中下载了模式物种酵母Saccharomyces cerevisiae、人类Homo sapiens和拟南芥Arabidopsis thaliana,以及与卵菌亲缘关系较近的硅藻Phaeodactylum tricornutum和假微型海链藻Thalassiosira pseudonana基因组中组蛋白H2A变异体的序列(表1),依据这些序列从BROAD INSTITUTE、DOE Joint Genome Institute、Ensembl Genomes和Pythium Genome Database搜索并下载致病疫霉菌Phytophthora infestans、大豆疫霉菌Phytophthora sojae、烁树猝死病菌Phytophhtora ramorum、角膜白斑病菌Albugo labachii、终极腐霉菌Pythium ultimum和寄生水霉菌Saprolegnia parasitica[17-20]6个卵菌基因组中的同源序列 (表1)。用模式物种的序列作为参比进行分析。利用SMART、Pfam、NCBI CD (Conserved Domains)-Search和InterProScan等在线程序对供试序列进行结构域预测与验证[21-23]。利用CPHmodels-3.2在线程序对致病疫霉组蛋白H2A变异体进行了三级结构预测[24]。
1.2.2 组蛋白H2A变异体序列比对和系统发育分析
通过利用在线程序ClusterW2[25],对供试物种的组蛋白H2A变异体保守结构域的氨基酸序列进行双重和多重序列比对。在双重序列比对中,gap的起始和延伸补偿值分别为10和0.1,相较而言,在多重序列比对中,gap的延伸补偿值为0.2。利用MEGA 5.22对序列比对后的结果进行聚类分析,用最大似然法构建聚类树[26]。Bootstrap值设定为1 000。
1.2.3 供试菌株RNA的提取
本试验中选取的供试菌株分别为2007年采自甘肃的致病疫霉A1交配型菌株MX5-1和YZ-6,以及来自美国的A2交配型菌株80787-94L[15]。收集菌株的无性生殖和有性生殖各阶段的菌丝体进行RNA提取。

表1 组蛋白H2A变异体基因信息表Table 1 Gene information for histone H2A variants

续表1
对于无性阶段,将MX5-1接种在黑麦番茄培养基上,共做150皿 (90 mm),然后置于18 ℃,黑暗培养10 d[15]。用无菌水冲洗收集游动孢子囊,将其中30皿的游动孢子囊悬浮液通过2 000 r/min离心8 min,收集游动孢子囊作为游动孢子囊阶段 (SP) 的试验样本。将剩余120皿的游动孢子囊悬浮液放置于4 ℃低温处理2-3 h诱导游动孢子形成和释放。2 000 r/min离心8 min收集其中30皿的游动孢子作为游动孢子阶段(ZO) 的试验样本。将另外30皿游动孢子悬浮液涡旋振荡40 s诱导游动孢子成为休止孢,然后再经过2 000 r/min离心8 min收集休止孢作为休止孢时期 (CY) 的试验样本。剩余的60皿游动孢子悬浮液用豌豆培养汁在120 r/min、18 ℃的条件下培养,其中30皿在培养3 h后用2 000 r/min离心8 min收集萌发的休止孢作为休止孢萌发阶段 (GC) 的试验样本,另外的30皿游动孢子悬浮液在培养48 h后用2 000 r/min离心8 min收集新鲜菌丝作为菌丝阶段 (MY) 的试验样本。对于有性阶段,用表面铺有一层孔径为0.4 μm的聚碳酸酯膜 (Millipore,Ireland,直径43 mm) 的黑麦番茄固体培养基平板(60 mm) 培养供试菌株,将YZ-6和80787-94L同时接种于一皿中,两菌丝块中间距离2 cm,处于18 ℃黑暗对峙培养诱导发生有性生殖,培养4 d、10 d和14 d后,分别刮取两菌丝块生长交界处的菌丝体和卵孢子,作为有性生殖过程中菌丝尖端膨大 (4DM),卵孢子形成 (10DM) 和卵孢子成熟 (14DM) 阶段的试验样本。同时,分别收集同样培养条件下,单独培养的菌龄4 d的YZ-6 (4D1) 和80787-94L (4D2) 菌丝体作为对照。
将这些样本收集后用吸水纸吸干水分,迅速用液氮研磨,然后于-80 ℃保存备用。样本的总RNA采用NucleoSpin RNA Plant Kit (740949,MACHEREY-NAGEL,Germany) 试剂盒进行提取,提取后用Thermo NanoDrop 2000分光光度计 (Wilmington,USA) 测量RNA的浓度和纯度并且用1.0%的琼脂糖凝胶电泳评估RNA质量。整个试验进行生物学重复一次。
1.2.4 表达水平测定与分析
首先针对致病疫霉3个组蛋白H2A变异体基因设计了特异性引物 (表2)。其中,PifH2A.X.1和PifH2A.X.2的序列相似度较高,通过序列比对,选取位于第70−180 bp序列差异较大的片段进行引物设计。利用SYBR实时荧光定量PCR的方法进行不同生长阶段基因表达水平测定。对于每一个样本,吸取1 μg总RNA用oligo (dT18) 作为引物,反转录酶M-MLV (TaKaRa Bio Inc.Shiga,Japan) 进行反转录反应获得cDNA。实时荧光定量PCR反应体系包括10 ng cDNA,引物0.2 μmol/L (终浓度),0.4 μL ROX Reference Dye II,10 μL SYBR Premix DimerEraser (Perfect Real Time) (TaKaRa Bio Inc. Shiga,Japan),加双蒸水补足到20 μL。利用ABI PRISM 7500 fast实时定量PCR仪 (Applied Biosystems,Foster City,SYBR CA,USA) 进行实时荧光定量PCR反应,Ct值反应程序为第一步95 30 s℃,第二步共40个循环,包括95 5 s℃,60 34 s℃;熔解曲线反应程序为95 ℃ 15 s,60 ℃ 1 min ,95 ℃ 15 s,用于检测实时荧光定量PCR引物的特异性。结果显示4对用于检测基因表达水平的引物均具有特异性。以延伸因子 (EF1,PITG_06722.1) 为内参基因,将实时荧光定量PCR结果数据进行分析处理,并绘制柱形图。
从表达数据库Gene Expression Omnibus DataSets得到Haas等利用基因芯片技术获得的致病疫霉侵染马铃薯不同时期的基因表达数据[20],据此分析了致病疫霉组蛋白H2A变异体基因在与寄主马铃薯互作过程中的表达情况。

表2 研究中使用的实时荧光定量PCR引物Table 2 Realtime qPCR primers used in this study
2 结果与分析
2.1 致病疫霉组蛋白H2A变异体的系统发育分析
通过在基因组数据中进行序列查找,以及与模式物种中组蛋白H2A序列比对分析,在致病疫霉基因组中发现存在编码2种组蛋白H2A变异体的基因,共3个,包括2个H2A.X (PITG_03881.1和PITG_03882.1) 和1个H2A.Z (PITG_02527.1)。同时,我们对同为卵菌的P. sojae、P. ramorum、Py. ultimum、S. parasitica和A. laibachii进行了H2A变异体基因的搜索,结果发现除了在S. parasitica中发现了常规组蛋白H2A和变异体H2A.Z,其他卵菌基因组中与致病疫霉相似,都只含有组蛋白H2A.X和H2A.Z两种变异体,没有发现常规组蛋白H2A (表1)。在酵母中也没有发现常规组蛋白H2A[27],推测是在进化过程中,组蛋白变异体H2A.X逐渐替代了常规H2A。
通过以人组蛋白变异体H2A.Bbd为外群进行系统发育分析,结果发现包括致病疫霉在内的卵菌中的组蛋白变异体H2A.X和常规体H2A,与模式物种酵母、拟南芥、人以及亲缘关系近的硅藻中的组蛋白变异体H2A.X和常规体H2A聚在一个大支上,而包括卵菌在内的不同物种中组蛋白变异体H2A.Z单独聚为一支(图1)。系统发育分析表明致病疫霉基因组中含有序列保守的组蛋白变异体H2A.X和H2A.Z,并且相对于同一物种中不同的组蛋白变异体,不同物种中的同类组蛋白变异体基因的相似性更高,说明不同物种中的同类组蛋白变异体有共同的起源。
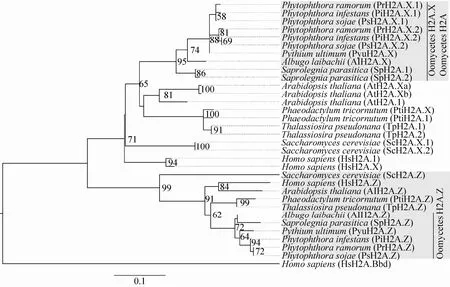
图1 致病疫霉组蛋白H2A变异体的ML系统发育树Fig. 1 Maximum likelihood phylogenetic tree of histone H2A variants of Phytophthora infestans. Bootstrap values less than 50 were hided.
2.2 致病疫霉组蛋白H2A变异体的序列结构分析
2.2.1 组蛋白H2A.X序列结构
致病疫霉基因组中含有2个编码组蛋白变异体H2A.X的基因:PITG_03881.1和PITG_03882.1。H2A.X.1编码一条长139个氨基酸的序列,H2A.X.2编码一条长140个氨基酸的序列。序列比对发现,这2个组蛋白H2A.X氨基酸序列相似度高达97.1%,主要差异位点在第16、19、24、39、47和49位 (图2)。
通过在蛋白数据库中进行序列比对,发现在已有三维结构解析的H2A.X中致病疫霉H2A.X.1和H2A.X.2与非洲爪蟾的H2A.X (PDB:5f99,chain G) 相似度最高,分别为78.2%和76.9%。以其为模板构建蛋白结构,发现H2A.X.1和H2A.X.2包含了5个alpha螺旋,这5个alpha螺旋在不同物种中十分保守 (图2)。
通过序列比对分析发现,致病疫霉组蛋白H2A.X.1和H2A.X.2中C端的丝氨酸 (S) 分别位于第136位和第137位,磷酸化保守位点为SQDY (图2),并且与硅藻C端磷酸化保守位点相同。而在酵母、拟南芥和人等的研究中发现,组蛋白H2A.X中保守的磷酸化位点位于序列C端SQEL/F/Y motif,其中的丝氨酸 (S) 可以发生磷酸化。尽管卵菌和模式物种中保守的磷酸化位点中有1个氨基酸不同,但谷氨酸 (E) 和天冬氨酸 (D) 同为酸性氨基酸,结构相似,仅差一个甲基。

图2 组蛋白H2A变异体H2A.X的氨基酸序列比对Fig. 2 Amino acid sequences alignments of histone H2A variants H2A.X. Highly conserved amino acids were showed with dark background. Structure of PiH2A.X.1 was predicted through modeling with H2A.X (PDB: 5f99, chain G) of Xenopus laevis, and illustrated as five alpha helixes. The phosphorylation motifs at C terminus were showed in black boxes.
2.2.2 组蛋白H2A.Z序列结构
致病疫霉基因组中含有1个编码组蛋白变异体H2A.Z的基因:PITG_02527.1。H2A.Z编码一条长136个氨基酸的序列。序列比对发现在已有三维结构解析的H2A.X中,致病疫霉组蛋白H2A.Z与人的H2A.Z (PDB:1f66,chain G)相似度最高 (87.5%),以其为模板构建蛋白结构,发现H2A.Z包含了5个alpha螺旋,序列比对结果显示这5个alpha螺旋在不同物种中十分保守 (图3)。

图3 组蛋白H2A变异体H2A.Z氨基酸序列比对Fig. 3 Amino acid sequences alignments of histone variants H2A.Z. Highly conserved amino acids were showed with dark background. Structure of PiH2A.Z was predicted through modeling with H2A.Z (PDB: 1f66, chain G) of human, and illustrated as five alpha helix.
2.3 编码致病疫霉组蛋白H2A变异体的基因表达谱分析
通过Realtime qPCR检测致病疫霉组蛋白H2A变异体在不同生长发育阶段的表达水平,以及致病疫霉侵染马铃薯不同阶段基因芯片的表达数据分析发现:致病疫霉组蛋白H2A.X.1在无性生长发育时期的游动孢子和休止孢阶段表达显著上调,而在游动孢子囊和萌发的休止孢阶段表达下调,在有性生殖阶段,随着卵孢子的形成表达水平逐渐下降,而在与寄主互作的过程中,表达水平在接种后2−3 d的时间段表达明显上调,4−5 d没有明显变化。结果表明H2A.X.1参与游动孢子、休止孢以及侵染早期相关基因的表达调控 (图4)。
与H2A.X.1相似,H2A.X.2在无性生长发育时期游动孢子和休止孢阶段表达上调,而在游动孢子囊和萌发的休止孢阶段表达显著下调,在有性生殖阶段初期和后期表达水平有所下降,而在与寄主互作的过程中,在接种后2 d表达略上调,其他阶段没有明显变化;结果表明H2A.X.2可能参与游动孢子、休止孢,有性生殖中期以及侵染早期相关基因的表达调控(图4)。
H2A.Z在无性生长发育时期游动孢子囊、游动孢子和休止孢阶段表达显著上调,而在萌发的休止孢阶段表达水平没有明显变化,有性生殖阶段,与H2A.X不同,随着卵孢子的形成表达水平逐渐上升,而在与寄主互作的过程中,随着侵染时间进程表达量逐渐降低;结果表明H2A.Z可能参与游动孢子囊、游动孢子、休止孢以及有性生殖后期相关基因的表达调控 (图4)。
分别针对致病疫霉生长发育阶段 (图4A和4B) 和与寄主互作阶段 (图4C) 过程中基因表达水平进行了研究,在Haas等测试致病疫霉在侵染寄主过程中的基因表达时,是以在不同培养基上的菌丝体作为对照进行分析,这一阶段的菌丝体可能包括了菌丝、游动孢子囊、游动孢子等结构,与图4A中无性生长发育阶段(MY、SP、ZO、CY和GC) 的样本可能有一定的重叠,但区别是前者为混合样本,后者是每一时期的单独测定结果,因此两者不具有可比性。同时,发现在图4C中,致病疫霉在不同培养基中生长,组蛋白H2A变异体基因的表达存在差异。相对于V8和RSA营养丰富的培养基而言,在寡营养的Pea培养基中组蛋白H2A变异体基因的表达相对较高,说明寡营养条件可能诱导组蛋白H2A变异体基因的表达。
研究还发现组蛋白H2A变异体3个基因的表达谱存在差异,说明不同的组蛋白H2A变异体可能具有特定的功能。由于mRNA的相对表达量,只能说明基因转录水平的相对变化,是否在蛋白质水平上也发生了相应的变化仍需要进一步研究。

图4 致病疫霉组蛋白H2A变异体基因在无性生长发育阶段 (A)、有性生殖阶段 (B) 和侵染阶段 (C) 的表达谱Fig. 4 Expression patterns of the histone H2A variants of Phytophthora infestans during asexual development (A), sexual reproduction (B) and infection stages (C). (A-B), The relative expression values were generated by realtime qPCR data from two biological repeats. (C) The relative expression values were generated by microarray expression data of GSE14480 retrieved from GEO DataSets.
3 结论
本研究通过基因序列比对,系统发育分析和基因表达数据分析发现致病疫霉等卵菌基因组中含有序列保守的组蛋白变异体H2A.X和H2A.Z,不存在变异体H2A.Bbd和MacroH2A。致病疫霉基因组中包含2个组蛋白变异体H2A.X、H2A.X.1和H2A.X.2,蛋白的C端含有保守的磷酸化位点SQDY。表达数据分析显示致病疫霉变异体H2A.X.1、H2A.X.2和H2A.Z可能参与调控特定的生长发育和侵染相关基因的表达。研究结果为进一步探索致病疫霉组蛋白H2A的功能建立了基础。
REFERENCES
[1] Luger K, Mäder AW, Richimond RK, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 1997, 389(6648): 251−260.
[2] Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nat Struct Biol, 2003, 10(11): 882-891.
[3] Ausió J, Abbott DW. The many tales of a tail: carboxyl-terminal tail heterogeneity specializes histone H2A variants for defined chromatin function. Biochemistry, 2002, 41(19): 5945−5949.
[4] Kimmins S, Sassone-Corsi P. Chromatin remodelling and epigenetic features of germ cells. Nature, 2005, 434(7033): 583−589.
[5] Talasz H, Helliger W, Sarg B, et al. Hyperphosphorylation of histone H2A.X and dephosphorylation of histone H1 subtypes in the course of apoptosis. Cell Death Differ, 2002, 9(1): 27−39.
[6] Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics, 2012, 7(10): 1098−1108.
[7] Larochelle M, Gaudreau L. H2A.Z has a function reminiscent of an activator required for preferential binding to intergenic DNA. EMBO J, 2003, 22(17): 4512−4522.
[8] Coleman-Derr D, Zilberman D. DNA methylation, H2A.Z, and the regulation of constitutive expression. Cold Spring Harb Symp Quant Biol, 2012, 77: 147-154.
[9] Abbott DW, Ivanova VS, Wang XY, et al. Characterization of the stability and folding of H2A.Z chromatin particles: implications for transcriptional activation. J Biol Chem, 2001, 276(45): 41945−41949.
[10] Jin JJ, Cai Y, Li B, et al. In and out: histone variant exchange in chromatin. Trends Biochem Sci, 2005, 30(12): 680−687.
[11] Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet, 2004, 20(7): 320−326.
[12] Tyler BM, Tripathy S, Zhang XM, et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science, 2006, 313(5791): 1261-1266.
[13] Fry W. Plant disease that changed the world Phytophthora infestans: the plant (and R gene) destroyer. Mol Plant Pathol, 2008, 9(3): 385-402.
[14] Han M, Liu G, Li JP, et al. Phytophthora infestans field isolates from Gansu province, China are genetically highly diverse and show a high frequency of self fertility. J Eukaryotic Microbiol, 2012, 60(1): 79−88.
[15] Guo LY, Zhu XQ, Hu CH, et al. Genetic structure of Phytophthora infestans populations in China indicates multiple migration events. Phytopathology, 2010, 100(10): 997-1006.
[16] Guo LY, Ko WH. Two widely accessible media for growth and reproduction of Phytophthora and Pythium species. Appl Environ Microbiol, 1993, 59(7): 2323−2325.
[17] de Bruijn I, Belmonte R, Anderson VL, et al. Immune gene expression in trout cell lines infected with the fish pathogenic oomycete Saprolegniaparasitica. Dev Comp Immunol, 2012, 38(1): 44-54.
[18] Lévesque CA, Brouwer H, Cano L, et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol, 2010, 11(7): R73, doi: 10.1186/gb-2010-11-7-r73.
[19] Kemen E, Gardiner A, Schultz-Larsen T, et al. Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol, 2011, 9(7): e1001094.
[20] Haas BJ, Kamoun S, Zody MC, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature, 2009, 461(7262): 393-398.
[21] Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res, 2015, 43(D1): D257-D260.
[22] Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database. Nucleic Acids Res, 2014, 42(D1): D222-D230.
[23] Marchler-Bauer A, Lu SN, Anderson JB, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res, 2011, 39(S1): D225-D229.
[24] Nielsen M, Lundegaard C, Lund O, et al. CPHmodels-3.0-Remote homology modeling using structure guided sequence profiles. Nucleic Acids Res, 2010, 38(S2): W576-W581, doi: 10.1093/nar/gkq535.
[25] Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics, 2007, 23(21): 2947-2948.
[26] Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol, 2011, 28(10): 2731-2739.
[27] Thatcher TH, Gorovsky MA. Phylogenetic analysis of core histone H2A, H2B, H3 and H4. Nucleic Acids Res, 1994, 22(2): 174-179.
(本文责编 陈宏宇)
Sequence analysis and expression patterns of histone H2A variants in Phytophthora infestans
Xiaowen Wang, and Liyun Guo
The Ministry of Agriculture Key Laboratory for Plant Pathology, Department of Plant Pathology, China Agricultural University, Beijing 100193, China
In eukaryotic cells, DNA is packaged inside the nucleus together with histones to form nucleosomes. Each histone molecule contains two of each core histone subunits H2A, H2B, H3 and H4. Among core histones, the H2A family is of great interest due to the high diversity of specialized variants. These variants have shown important role in critical cellular processes. Epigenetic mechanism in oomycetes is barely known. Phytophthora infestans is a severe pathogen and amodel species in oomycetes. In this study, we studied the sequence and expression pattern of H2A variants of P. infestans through genome search, sequences alignment, phylogenetic analysis and realtime qPCR detection. P. infestans contains conserved genes encoding histone variants H2A.X.1, H2A.X.2 and H2A.Z, and these genes have specific expression patterns during development and infection stages. Our datasets provide useful inputs to help explore the epigenetic mechanisms of oomycetes.
March 18, 2016; Accepted: April 28, 2016
Liyun Guo. Tel/Fax: +86-10-62733032; E-mail: ppguo@cau.edu.cn, ppguoly@126.com
oomycetes, late blight pathogen, H2A.X, H2A.Z, phylogeny, expression pattern
Supported by: National Natural Science Foundation of China (No. 30270862).
国家自然科学基金 (No. 30270862) 资助。
猜你喜欢
哈尔滨轴承(2021年1期)2021-07-21
绵阳师范学院学报(2021年5期)2021-05-28
小学科学(2020年11期)2020-03-04
河北大学学报(自然科学版)(2020年1期)2020-01-15
广东农业科学(2017年5期)2017-08-29
渔业致富指南(2016年12期)2016-11-11
科学大众(中学)(2015年9期)2015-10-12
少儿科学周刊·少年版(2015年3期)2015-07-07
天津诗人(2014年4期)2014-11-14
华东师范大学学报(自然科学版)(2014年6期)2014-02-27
