
Tetrahydrohyperforin (IDN5706) targets the endoplasmic reticulum for autophagy activation: potential mechanism for Alzheimer’s disease therapy
2016-12-02 02:29AlexisGonzález,VivianaA.Cavieres,NibaldoC.Inestrosa等
中国神经再生研究(英文版) 2016年2期
PERSPECTIVE
Tetrahydrohyperforin (IDN5706) targets the endoplasmic reticulum for autophagy activation: potential mechanism for Alzheimer’s disease therapy
Alzheimer’s disease (AD) is the most common form of dementia worldwide among the older population. To date, there is no therapy to stop the destruction of brain cells and all the available treatments only compensate for the loss of synaptic transmission, thus resulting in marginal benefits to patients.
To prevent brain cell loss, we need to identify and understand the exact causes of the disease. One of the most studied theories is the amyloid hypothesis on either the overproduction of the amyloid-β (Aβ) peptide species, or impairment of the mechanism that usually clears it from specific regions of the brain, or both, triggers the disruption and damage of brain cells. Aβ contributes to the formation of intracellular tangles made up of a protein called tau, that together with extracellular Aβ contributes to the symptoms of dementia, and to the development of AD. Mechanistically, Aβ is derived from the sequential proteolysis of a large transmembrane protein called amyloid precursor protein (APP). Cleavage of APP by the β-site APP cleaving enzyme 1 (γ-secretase) generates the C-terminal fragment C99 that can be subsequently cleaved by γ-secretase to produce Aβ (Bustamante et al., 2013). Despite the significant effort in the design of a treatment to prevent Aβ formation by interfering with β- and γ-secretase activities, these strategies have failed due to several side effects caused by the inhibition of the proteolytic processing of a variety of substrates, other than APP. In contrast, a recent study reduces the possibility of Aβ formation and promising strategy explored a diff erent possibility, which is by enhancing the clearance of APP and/or C99 (Tian et al., 2011). Our recent work supported the aforementioned conclusion and highlighted the crucial role of the endoplasmic reticulum (ER) in the process (Cavieres et al., 2015). Here we propose that positive regulators of ER autophagy-related responses, such as tetrahydrohyperforin, are promising niches for the discovery of future therapies against AD.
Tetrahydrohyperforin prevents cognitive deficits in an AD model: possible mechanism links clearance of APP by the activation of autophagy at the ER: A recent study from our group using a neuroprotective semisynthetic derivative of hyperforin, the active molecule in the St John´s Wort plant (Hypericum perforatum) called tetrahydrohyperforin (IDN5706), has demonstrated that this compound prevents the neuropathological changes in a mouse model of AD. Specifically, it induces long-term potentiation (LTP) and prevents AD-associated loss of spatial memory, reduces tau hyperphosphorylation, and decreases Aβ peptide levels (Inestrosa et al., 2011). In an effort to elucidate the molecular mechanism that could explain the beneficial effects of this compound, we recently showed that IDN5706 targets the ER for autophagy activation, triggering entry of immaturely glycosylated, newly-synthesized APP (iAPP) in ER-associated structures, which favors its degradation by Atg5-dependent autophagy, leading to inhibition of Aβ peptide formation (Cavieres et al., 2015).
Autophagy, a major lysosomal degradative pathway, has been extensively studied in age-related neurodegenerative disorders, such as AD, due to its strong connection between aging and the progressive deterioration in the proteostatic capacity of the brain. Although, induction of autophagy delays aging, reduces the risk of neurodegenerative disorders and neuronal dysfunction in animal models, the underlying mechanisms still remain poorly understood. In this context, our recent findings with the neuroprotective compound IDN5706 revealed that the ER could be a major regulator for autophagy activation, necessary to promote the clearance of APP upon perturbations in its glycosylation (Cavieres et al., 2015), strongly supporting the hypothesis that autophagy could play a key role in quality control of the ER.
The ER connects with lysosomes in alert conditions: Several recent studies support the idea of a functional crosstalk between the ER and lysosomes. While constitutive pathways play a well-known role in controlling ER homeostasis (Figure 1), a growing number of reports indicated that ER activates a variety of cellular responses under conditions that disrupt ER proteostasis, such as perturbations in glycosylation, triggering rescue responses necessary to mitigate cellular damage (Figure 2). Taken together, this indicates that the ER plays a crucial role in quality control (ERQC) of newly synthesized proteins, a key step for the maintenance of cellular homeostasis. For instance, whereas ERQC “proof-reads” native proteins before delivering them into the secretory pathway, ER-associated degradation (ERAD), a constitutive mechanism, delivers proteins for cytosolic degradation by the proteasome through the well-defined ERAD pathway, a response that has been confirmed to participate in the regulation of APP and C99 cellular levels (Bustamante et al., 2013). Another well-known characterized pathway is the unfolded protein response (UPR) that induces the expression of a variety of chaperones and proteins in response to ER stress that aims to recover ER status and proteostasis balance. Additionally, ER stress conditions induce a specific type of autophagy that selectively targets ER membranes. This process is termed ER-Phagy and plays a crucial role in ERQC from yeast to humans (Khaminets et al., 2015). It participates in the removal of damaged proteins and redundant parts of the ER, thus contributing to the maintenance of ER homeostasis and neuronal viability (Khaminets et al., 2015). Interestingly, carbamazepine (CBZ), an autophagy-enhancing drug, has been proven to enhance the degradation of mutant α1-antitrypsin (ATZ), a protein that is otherwise abnormally retained in the ER of liver cells. CBZ-induced clearance of mutant ATZ is dependent on Atg5, a protein that has been shown to be recruited to ER membranes to control autophagosomal formation and ER-Phagy. Similarly to CBZ, we recently showed that degradation of iAPP induced by IDN5706 treatment is also dependent on Atg5 levels (Cavieres et al., 2015). Finally, another autophagy-like mechanism, distinct from macroautophagy, is known as ERAD tuning, a pathway involved in the formation of ER membrane vesicles decorated by nonlipidated LC3 at their cytosolic face, called EDEMosomes. These membranes segregate a variety of ERAD factors for further degradation in lysosomes to avoid premature interruption by early degradation through ERAD. A variety of viruses, classical ER stressors, evade immunological surveillance by hijacking host cell ERAD machinery, which allows for efficient viral production and persistency. In this regard, it has been described that Coronaviruses (CoV) sequester ERAD regulators and EDEMosomes for their own replicative process, affecting ERAD and the ERAD tuning pathway. Our recent findings with IDN5706 indicated that the reduction of the ER degradation enhancer, mannosidase alpha-like 1 (EDEM1), a key ERAD factor, could also function as a cellular response to assure degradation of immaturely glycosylated proteins by the lysosomes (ER-phagy), instead of by the proteasome (ERAD), a hypothesis still under investigation. Interestingly, overexpression of a key protein of ER-Phagy recently identified, FAM134B (Khaminets et al., 2015) resulted in ER fragmentation and lysosomal degradation. In contrast, downregulation or expression of mutant FAM134B proteins resulted in sensitization of sensory neurons to neurodegeneration (Khaminets et al., 2015). Altogether, these results suggest that ER-autophagy related responses could play unexplored functions in ERQC, which regulates levels of proteins such as APP under normal and pathological conditions. This could be crucial for avoiding deterioration of neuronal function and the development of AD.
Autophagy activation as a helper for healthy brain function: To date, increasing evidence supports a key role of autophagy for healthy brain function. The most compelling findings show that expression of Atg5 has been manipulated in animal models. Atg5-deficient mice displayed abnormal accumulation of cytoplasmic ubiquitin-positive inclusions, a phenotype that correlates with progressive neurodegeneration in different regions of the brain, including the cerebral cortex and hippocampus (Hara et al., 2006). On the contrary, overexpression of Atg5 in mice showed a significant increase in LC3 levels in the brain, a feature indicative of autophagy activation that has been proven to extend lifespan (Pyo et al., 2013). In addition, studies with the mTOR signaling pathway, a serine/threonine kinase that is recognized as a major negative regulator of autophagy in the nervous system, support the same conclusion. In this context, reduction of mTOR signaling by chronic administration of rapamycin has been demonstrated to extend lifespan in model organisms and confers protection against AD (Harrison et al., 2009). Our recent findings indicate that mTOR inhibition is also implicated in the activation of autophagy by IDN5706, suggestingthat regulation of ERQC by ER autophagy-related mechanisms could also be regulated by mTOR signaling, a possibility that remains unexplored at present. In this respect, it will be interesting to explore whether inducers of ER autophagy-related mechanisms, including IDN5706, can extend lifespan in animal models.
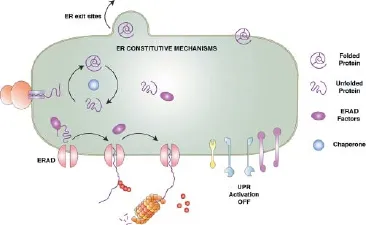
Figure 1 Summary of the constitutive pathways at the endoplasmic reticulum (ER) that control proteostasis.
AD: the hypothesis of an autophagy-related disease: To date, there is evidence indicating that AD brains present wide alterations of autophagy. Indeed, the observation of a massive accumulation of autophagic vacuoles (AVs) in abnormally swollen neurites was the first evidence that supported this conclusion. In addition, it has been demonstrated that enhancing lysosomal function ameliorates the cognitive deficits in animal models of AD, indicating that autophagy failure contributes to AD pathogenesis (Boland et al., 2008). However, it is still unclear what the real contribution of autophagy to the development of AD is. Is it triggered because of a dysfunctional autophagy pathway or due to the accumulation of specific substrates that are not efficiently eliminated? In any case, these possibilities seem to be implicated in the destruction of the neuronal cells in AD brains. Interestingly, recent evidence supports the role of autophagy in the clearance of APP and C99, two unexpected autophagy substrates (Tian et al., 2011; Cavieres et al., 2015). Moreover, our recent work opens the opportunity to explore the contribution of the ER in the activation of autophagy and its functional role in the early clearance of APP and control of Aβ formation. We propose that IDN5706 is an attractive molecule to identify key molecular players necessary for the activation of these early responses, which can lead to the development of drugs to treat AD.
This work was supported by FONDECYT-1130929 (PB) and PB-Conicyt (No 12/2007) to NCI. AG is supported by CONICYT#21110746, MECESUPAUS1203 and DIDUAChD#201303. VC is supported by CONICYT#21151194 fellowship.
Alexis González#, Viviana A. Cavieres#, Nibaldo C. Inestrosa, Patricia V. Burgos*
Instituto de Fisiología, Facultad de Medicina; Centro Interdisciplinario de Estudios del Sistema Nervioso (CISNe), Universidad Austral de Chile, Valdivia, Chile (González A, Cavieres VA, Burgos PV) Centro de Envejecimiento y Regeneración (CARE); Centro UC Síndrome de Down; Pontificia Universidad Católica de Chile, Santiago, Chile/Center for Healthy Brain Ageing, School of Psychiatry, Faculty of Medicine,
University of New South Wales, Sydney, Australia/Centro de Excelencia en Biomedicina de Magallanes (CEBIMA), Universidad de Magallanes, Punta Arenas, Chile (Inestrosa NC)
*Correspondence to: Patricia V. Burgos, Ph.D., patricia.burgos@uach.cl.
#Both authors contributed equally to this paper.
Accepted: 2015-12-18

Figure 2 Summary of the endoplasmic reticulum (ER) stress responses.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28:6926-6937.
Bustamante HA, Rivera-Dictter A, Cavieres VA, Munoz VC, Gonzalez A, Lin Y, Mardones GA, Burgos PV (2013) Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS One 8:e83096.
Cavieres VA, Gonzalez A, Munoz VC, Yefi CP, Bustamante HA, Barraza RR, Tapia-Rojas C, Otth C, Barrera MJ, Gonzalez C, Mardones GA, Inestrosa NC, Burgos PV (2015) Tetrahydrohyperforin inhibits the proteolytic processing of amyloid precursor protein and enhances its degradation by Atg5-dependent autophagy. PLoS One 10:e0136313.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885-889.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392-395.
Inestrosa NC, Tapia-Rojas C, Griffith TN, Carvajal FJ, Benito MJ, Rivera-Dictter A, Alvarez AR, Serrano FG, Hancke JL, Burgos PV, Parodi J, Varela-Nallar L (2011) Tetrahydrohyperforin prevents cognitive deficit, Abeta deposition, tau phosphorylation and synaptotoxicity in the APPswe/PSEN1DeltaE9 model of Alzheimer’s disease: a possible effect on APP processing. Transl Psychiatry 1:e20.
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522:354-358.
Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK (2013) Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4:2300.
Tian Y, Bustos V, Flajolet M, Greengard P (2011) A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. Faseb J 25:1934-1942.
10.4103/1673-5374.177728 http://www.nrronline.org/
How to cite this article: González A, Cavieres VA, Inestrosa NC, Burgos PV (2016) Tetrahydrohyperforin (IDN5706) targets the endoplasmic reticulum for autophagy activation: potential mechanism for Alzheimer´s disease therapy Neural Regen Res 11(2):242-243.
- 中国神经再生研究(英文版)的其它文章
- Tissue-type plasminogen activator is a modulator of the synaptic vesicle cycle
- Impaired consciousness caused by injury of the lower ascending reticular activating system: evaluation by diffusion tensor tractography
- Considering calcium-binding proteins in invertebrates: multi-functional proteins that shape neuronal growth
- Cardiovascular dysfunction following spinal cord injury
- Practical application of the neuroregenerative properties of ketamine: real world treatment experience
- Exergames: neuroplastic hypothesis about cognitive improvement and biological effects on physical function of institutionalized older persons