
Exploring a need for improved preclinical models of post-stroke depression
2016-12-02 06:04:21NitishMittal,PatriciaD.Hurn,TimothySchallert
中国神经再生研究(英文版) 2016年4期
PERSPECTIVE
Exploring a need for improved preclinical models of post-stroke depression
Depression is one of the most common neuropsychiatric disorders associated with stroke. One in three patients who suffer from a stroke, report having depression-like symptoms. The mean prevalence for major and minor depression amongst stroke in patients is 21.6% and 20.0% respectively; the mean prevalence for outpatients is 24.0% and 23.9%. Increased severity of post-stroke depression has been associated with increased cognitive impairment and mortality. It is also considered to be one of the strongest correlates of impairment in activities of daily living after stroke. Whether the severity of depression impacts the degree of impairment, or vice versa, is not yet fully understood. Initially, post-stroke depression was thought to be a psychological response to the disability produced by stroke; however, studies in orthopedic patients with similar levels of disability, but a lower prevalence of depression, provide some evidence against such purely psychological explanations of post-stroke depression (Robinson and Spalletta, 2010).
Depression has also been shown to slow post-stroke recovery and negatively impact functional outcomes. Indeed, depressed patients often have poorer results on the Modified Rankin Scale up to 12 months after stroke, as compared to non-depressed patients (Wulsin et al., 2012). Furthermore, a 10-year follow-up study in 37 stroke patients showed that the mortality rate for depressed patients was 70% by year 10, as compared to 41% for non-depressed patients. Moreover, patients that show remission from depression also show greater recovery on the Johns Hopkins Functioning Inventory at 3 and 6 months of follow-ups after stroke (Flaster et al., 2013). These studies highlight an important need for understanding the pathobiology of post-stroke depression. Identifying the mechanisms that contribute to depression, and subsequently impact patient recovery after stroke, will help us design better treatments and improve patient quality of life.
Preclinical studies of post-stroke depression: Clinical studies in patients are a great way to identify problems that impact patient care; however, in order to study the neural mechanisms behind these pathologies, it is critical to replicate these results in preclinical experiments. Many factors that are thought to underlie post-stroke depression have been identified; these include: neurotrophin signaling, genomic signatures, changes in synaptic plasticity, and neuroinflammation. For more details regarding these mechanisms, please refer to the review by Loubinoux et al. (2012). Several preclinical models of post-stroke depression have been used to shed light on these mechanisms. For instance, induction of depression by exposing rats to chronic mild stress after stroke has been shown to reduce hippocampal neurogenesis, decrease the expression of serotonin 1A receptor in the dentate gyrus, and in turn result in worse functional outcomes after stroke. Similarly, social isolation after stroke can also produce post-stroke depression in rodents, and has been associated with greater infarct volume, decreased BDNF levels, as well as increased serum expression levels of the inflammatory cytokine interleukin 6. Furthermore, these symptoms can be reversed by pair housing the animals. Social stress and corticosterone-induced stress can also increase infarct volume after stroke, and this can be reversed with the administration of glucocorticoid receptor antagonists. Other studies have shown that chronic stress increases heart rate, and subsequently impairs endothelial vascular function, upregulates lipid hydroperoxides in the brain, and increases lesion size after stroke. Moreover, these symptoms can be reversed by decreasing heart rate via the administration of ivabradine, a vascular potassium channel inhibitor (Kronenberg et al., 2014).
These studies have confirmed the clinical findings regarding the comorbidity between depression and stroke by demonstrating that depression and stress can indeed worsen the outcome of ischemic stroke in rodents. Furthermore, they have also identified several physiological substrates that may have a potential role in worsening stroke induced damage, and subsequently, negatively affecting functional recovery after stroke. However, these modelsrely on additional stimuli to induce stress or depression in subjects, which may not be directly comparable to the post-stroke depression that occurs directly as a result of stroke in many human patients. While it is possible that patients who suffer from a stroke may feel socially isolated, which could in turn lead to depression, this is more likely in cases of late onset of post-stroke depression. This is indeed an important risk factor one year after stroke (Whyte and Mulsant, 2002), but not immediately after stroke as seen in the aforementioned animal studies.
The tests commonly used to assess mood disorders in rodents include Porsolt’s forced swim test, the tail suspension test, the sucrose consumption test, the novelty-suppressed feeding test, and the elevated plus maze test. Porsolt’s forced swim test and the tail suspension test are thought to be indicators of despair and use the animal’s latency to ‘give up’ as a measure of depression. The sucrose consumption test is considered a rodent marker of anhedonia, which reflects an inability to enjoy otherwise palatable substances. The novelty-suppressed feeding task and the elevated plus maze tests, on the other hand, are primarily used as measures of anxiety, and not depression. Although these assays have served as useful tools for screening anti-depressants, one objection is that these tests typically respond to acute administration of anti-depressants; whereas it takes weeks of treatment with the same drugs for patients to recover from clinical depression. For in-depth discussion of other problems with currently used preclinical models of depression please see the following reviews by Nestler and Berton (Nestler et al., 2002; Berton et al., 2012).
Few studies have been able to show depression-like symptoms in rodents after the induction of stroke alone, and when they have, the results have been inconsistent. Some studies have shown that only left hemispheric infarction produced anxiety or depression-like behaviors in rodents, as measured by the Porsolt’s forced swim test, the sucrose consumption test, the tail suspension test, and the novelty suppressed feeding test; whereas right hemispheric infarctions resulted in hyperlocomotor activity in both the open field and running wheel tasks (Robinson, 1979). Other studies have shown that right hemispheric infarctions can indeed produce depression and anhedonia-like symptoms in rodents, as measured by the sucrose consumption test (Craft and DeVries, 2006). Yet other studies have found noeffect of right hemispheric infarctions on locomotor activity (Kronenberg et al., 2014; O’Keefe et al., 2014). One potential explanation for such mixed results may be that only a subset of stroked animals suffer from post-stroke depression, as is the case with human subjects, and therefore the prevalence of depression may not become apparent with the small group sizes that are typical of preclinical studies.
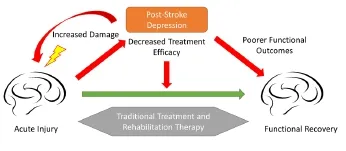
Figure 1 Schematic representation of the effects of post-stroke depression on recovery after stroke.
A potential role for the long jump test: Currently used rodent models of depression focus primarily on two out of the nine symptoms of depression (namely despair and anhedonia) delineated by the Diagnostic and Statistical Manual of Mental Disorders (DSM). More assays are needed to account for the remaining symptoms that include: change in sleep, loss of appetite, increased irritability, low confidence, and feelings of worthlessness. Future studies would need to utilize a battery of assays and aim to identify changes in more than one symptom to properly measure the prevalence of depression in rodent models of stroke. Our lab recently described a novel long jump assay in mice, which may serve as an addition to such an inventory of rodent tests for depression (Mittal et al., 2015).
The long jump test was initially designed to be a measure of hindlimb functionality after stroke. We hypothesized that the induction of an ischemic stroke will weaken the hindlimbs of mice, and hamper their ability to successfully jump from a beam into their home cage. We were surprised to find that a stroke induced by a 45-minute MCAO procedure was unable to produce sufficient deficits in jump success rate. The post-stroke animals were able to jump as successfully as the sham animals, but did appear more hesitant to jump. This hesitation was reflected in their increased latency to jump after stroke, which was nearly three times higher than baseline levels. We then postulated that the increase in jump latency may be a reflection of a psychological or cognitive change produced by stroke in these animals. In order to test this idea, we first used the elevated plus maze test to assess any potential increases in anxiety after stroke. Next we tested the effect of treatment with a selective serotonin reuptake inhibitor (SSRI), fluoxetine, on the long jump test. We were unable to attribute the deficit in jump latency to either of these possibilities. However, in this study, we did not use a chronic treatment regimen of fluoxetine, which may have been more effective in treating depression and potentially reversing the increase in jump latency. We also did not compare the increase in jump latency with results from another test of depression, which may have shed some light on the nature of this phenotype.
It is possible that the increase in latency to jump may be caused by a potential impairment in the visual field resulting from the ischemic insult. The increase in latency could also be a result of cognitive impairment due to the ischemic injury. Therefore, we felt that further characterization of this test as a measure of anxiety or depression would be better suited in other rodent models of anxiety or depression, such as chronic stress, social defeat or learned helplessness. These models are not encumbered by the sensorimotor deficits seen in stroked mice, and can be used to test longer anti-depressant treatment regimens. Nonetheless, the sensitivity of this test in detecting a change in latency after stroke is promising. Once the nature of this change is understood, the long jump test may serve as an important part of the inventory of preclinical assays designed to detect psychological deficits in stroked mice.
Conclusion: Depression affects a large subset of stroke patients, yet there is little insight into the underlying neurobiological mechanisms of post-stroke depression. The use of preclinical models to understand the nature of this pathology will help guide future clinical studies in this field. It is apparent that post-stroke depression interferes with functional recovery after stroke (Figure 1), yet the mechanisms behind this phenomenon are not well understood. In particular, recent clinical studies have highlighted the beneficial effects of anti-depressants, specifically SSRIs, on recovery after stroke. These effects cannot simply be attributed to the anti-depressant properties of these drugs, as they have been shown to improve recovery even in non-depressed patients (Chollet et al., 2013). The beneficial effects of SSRIs are thought to be mediated by their ability to regulate cortical excitability and synaptic reorganization that are important for recovery after stroke. Future pre-clinical studies will need to identify the specific mechanisms through which monoaminergic anti-depressants impact synaptic reorganization. Such studies will also be important in highlighting the ways in which the effects of SSRIs on motor recovery differ from their effects on depression. While the literature on this subject is rather limited at this time, current studies highlight an important need for further investigation. As we find new and improved ways to detect post-stroke depression symptoms in rodents, we will be better equipped to explore its precise role on neurological damage and recovery.
Nitish Mittal*, Patricia D. Hurn, Timothy Schallert
College of Pharmacy, University of Texas at Austin, Austin, TX, USA (Mittal N)
Departments of Neuroscience (Hurn PD) and Psychology (Schallert T), University of Texas at Austin, Austin, TX, USA University of Texas System, Austin, TX, USA (Hurn PD)
*Correspondence to: Nitish Mittal, mittal.n@utexas.edu.
Accepted: 2016-03-03
orcid: 0000-0002-4934-3157 (Nitish Mittal)
Berton O, Hahn CG, Thase ME (2012) Are we getting closer to valid translational models for major depression? Science 338:75-79
Chollet F, Acket B, Raposo N, Albucher JF, Loubinoux I, Pariente J (2013) Use of antidepressant medications to improve outcomes after stroke topical collection on stroke. Curr Neurol Neurosci Rep 13.
Craft TKS, DeVries AC (2006) Role of IL-1 in poststroke depressive-like behavior in mice. Biol Psychiatry 60:812-818.
Flaster M, Sharma A, Rao M (2013) Poststroke depression: a review emphasizing the role of prophylactic treatment and synergy with treatment for motor recovery. Top Stroke Rehabil 20:139-150.
Kronenberg G, Gertz K, Heinz A, Endres M (2014) Of mice and men - modeling post-stroke depression experimentally. Br J Pharmacol 171:4673-4689.
Loubinoux I, Kronenberg G, Endres M, Schumann-Bard P, Freret T, Filipkowski RK, Kaczmarek L, Popa-Wagner A (2012) Post-stroke depression: Mechanisms, translation and therapy. J Cell Mol Med 16:1961-1969.
Mittal N, Pan J, Palmateer J, Martin L, Pandya A, Kumar S, Ofomata A, Hurn PD, Schallert T (2015) So you think you can jump? A novel long jump assessment to detect deficits in stroked mice. J Neurosci Methods 256:212-219.
Nestler EJ, Gould E, Manji H (2002) Preclinical models: Status of basic research in depression. Biol Psychiatry 52:503-528.
O’Keefe LM, Doran SJ, Mwilambwe-Tshilobo L, Conti LH, Venna VR, Mc-Cullough LD (2014) Social isolation after stroke leads to depressive-like behavior and decreased BDNF levels in mice. Behav Brain Res 260:162-170.
Robinson RG (1979) Differential behavioral and biochemical effects of right and left hemispheric cerebral infarction in the rat. Science 205:707-710.
Robinson RG, Spalletta G (2010) Poststroke depression: a review. Can J Psychiatry 55:341-349.
Whyte EM, Mulsant BH (2002) Post stroke depression: Epidemiology, pathophysiology, and biological treatment. In: Biological Psychiatry, pp253-264.
Wulsin L, Alwell K, Moomaw CJ, Lindsell CJ, Kleindorfer DO, Woo D, Flaherty ML, Khatri P, Adeoye O, Ferioli S, Broderick JP, Kissela BM (2012) Comparison of two depression measures for predicting stroke outcomes. J Psychosom Res 72:175-179.
10.4103/1673-5374.180736 http://www.nrronline.org/
How to cite this article: Mittal N, Hurn PD, Schallert T (2016) Exploring a need for improved preclinical models of post-stroke depression. Neural Regen Res 11(4):561-562.

- 中国神经再生研究(英文版)的其它文章
- Gait deterioration due to neural degeneration of the corticoreticular pathway: a case report
- Complement components of nerve regeneration conditioned fluid influence the microenvironment of nerve regeneration
- Electrical stimulation of dog pudendal nerve regulates the excitatory pudendal-to-bladder reflex
- Supplementary motor area deactivation impacts the recovery of hand function from severe peripheral nerve injury
- Combined use of Y-tube conduits with human umbilical cord stem cells for repairing nerve bifurcation defects
- Senegenin inhibits neuronal apoptosis after spinal cord contusion injury