
2,6-位取代的BODIPY染料敏化剂的合成及其性能
2016-12-01 01:32周伟男赵鸿斌蔡卓弟廖俊旭彭飞严文杰阳年发湘潭大学化学学院湘潭405东莞理工学院化学与环境工程学院东莞5808东莞市化工学会东莞5808
无机化学学报 2016年1期
周伟男 赵鸿斌 蔡卓弟 廖俊旭, 彭飞 严文杰 阳年发(湘潭大学化学学院,湘潭405)(东莞理工学院化学与环境工程学院,东莞5808)(东莞市化工学会,东莞5808)
2,6-位取代的BODIPY染料敏化剂的合成及其性能
周伟男1,2赵鸿斌*,2蔡卓弟3廖俊旭*,2,3彭飞1,2严文杰2阳年发*,1
(1湘潭大学化学学院,湘潭411105)
(2东莞理工学院化学与环境工程学院,东莞523808)
(3东莞市化工学会,东莞523808)
设计合成了3种新型基于氟化硼络合二吡咯甲川(BODIPY)衍生物的D-π-A型光敏染料CB1~CB3,其结构为:BODIPY核为桥联基团、N-苯基咔唑以不同位点连接BODIPY的2-位作为电子给体单元、氰乙酸连接于BODIPY的6-位作为电子受体单元。运用1H,13C NMR以及MALDI-TOF-MS对所合成的染料CB1~CB3进行了结构表征和确证。对3种染料CB1~CB3进行了UV-Vis和荧光等光物理特性、电化学行为、光伏性能研究,并通过DFT理论计算深入研究了其分子的几何结构与性能之间的关系。3种染料的吸收光谱主要位于420~600 nm波段,LUMO能级在3.7 eV左右,HOMO能级在5.2 eV左右。N-苯基咔唑2-位与BODIPY相连的CB2,因更平整的分子结构和更为适合的共轭长度,具有更为优良的光谱吸收和分子内电荷迁移性能,因而表现出良好的光伏性能:在AM1.5(100 mW·cm-2)的光强下,CB2敏化电池的开路电压(Voc)为550 mV,电流密度(Jsc)为3.71 mA· cm-2,填充因子(FF)为0.73,总光电转换效率(PCE)为1.49%。
染料敏化太阳能电池;有机染料;光伏性能;BODIPY
自1991年瑞士的Grätzel小组[1]首次采用联吡啶钌作为染料与纳米多孔TiO2薄膜制备了染料敏化太阳能电池(DSSCs)以来,染料敏化纳米晶TiO2太阳能电池就凭借低制备成本和较高的光电转换效率,成为近20年来太阳能光电转换领域的研究热点[2-3]。光敏染料主要包含金属配合物和纯有机染料。目前,以多吡啶钌配合物为主的金属配合物因其良好稳定性和较高的能量转换效率,得到了广泛而深入的研究[4-5]。但金属配合物的制备过程较复杂、成本较高、且要用到贵重金属,因此,开发低成本、高性能的纯有机染料敏化剂逐渐受到科研工作者的重视[6]。近年来,纯有机染料敏化剂因其原料成本低廉、结构设计灵活、易合成、易提纯等优点,已经成为当前DSSCs研究的热门方向[7-12]。目前已报道的有机染料主要为电子给体-共轭π桥-电子受体(D-π-A)结构,该结构中π桥的引入有利于分子内的电子迁移(ICT)和激发态的电子分离[13],已成为有机染料的首选结构。
氟硼络合二吡咯甲川(BODIPY)衍生物是一类良好的光敏染料[14-16],具有良好的稳定性、可修饰性好、高的摩尔消光系数(1×105L·mol-1·cm-1)和较高的氧化电位,其应用于染料敏化太阳能电池已有文献报道[17-22]。BODIPY的光谱吸收相对较窄,然而其良好的可修饰性可以克服这一缺点,实现光谱吸收的红移和拓宽[23-24],有利于提高电池的Jsc。目前基于BODIPY衍生物染料敏化太阳能电池材料的已经获得了较高的光电转换效率[25],展现出了良好的应用前景。咔唑衍生物是另一类具有良好光电性质的化合物,其具有强的光谱吸收和光谱发射性能、很高的空穴传输能力以及较宽的带隙,在光电材料领域有广泛的应用[26-27]。鉴于两类材料突出的光电性质,因此,将BODIPY与咔唑类化合物结合起来,应用于染料敏化太阳能电池引起了我们的研究兴趣。
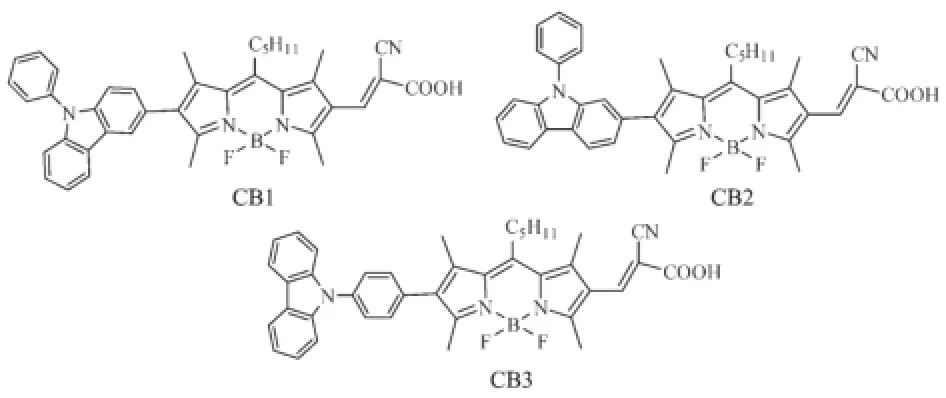
图式1 BODIPY染料敏化剂CB1~CB3的结构Scheme 1 Structures of BODIPY dye sensitizers (CB1~CB3)
本文以BODIPY为桥联结构,N-苯基咔唑不同取代位置连接BODIPY桥作为给体单元,氰乙酸为受体单元,设计合成3种新型D-π-A结构的BODIPY染料敏化剂CB1~CB3(图式1),并对其光物理性质、电化学行为和光伏性能进行研究。考察该类分子结构-性能之间的关系,即N-苯基咔唑2-位、3-位及苯基4-位连接方式对分子性能的影响规律;探索这种咔唑-BODIPY共轭结构在光电材料领域中的应用前景,期望开发优良的光电材料。
1实验部分
1.1试剂与仪器
论文中所用到的试剂均为市售分析纯或化学纯试剂,二氯甲烷和N,N-二甲基甲酰胺用氢化钙处理,四氢呋喃用钠丝回流。化合物1a[28]、1b[29]、1c[30]和2[25]参照文献合成,起始原料2,4-二甲基吡咯购于成都贝斯特试剂有限公司,咔唑、四(三苯基膦)钯、二茂铁和哌啶购于国药试剂,2-溴咔唑购于濮阳惠成电子材料有限公司,其他所用试剂均购于阿拉丁试剂有限公司。
Bruker 600 MHz核磁共振仪(瑞士Bruker公司); Bruker 400 MHz核磁共振仪(瑞士Bruker公司);Bruker ultraflex-Ⅱ型飞行时间质谱仪(瑞士Bruker公司);GCMS-QP2010气质联用仪(日本Shimadzu公司);UV3100紫外-可见分光光度计(日本Shimadzu公司);CHI660E电化学工作站(上海辰华仪器有限公司);F-4500荧光分光光度计(日本Hitachi公司);J-V测试使用的记录仪为Keithley 2602 Source meter,白光光源为AM1.5G(100 mW·cm-2)太阳光模拟器,电池的有效面积为0.196 cm-2。
1.2实验过程
1.2.1电池的制作
1.2.1.1光阳极制作
1.2.1.1.1 FTO的清洗
首先将FTO玻璃划成10 cm×3.8 cm的大小,并在导电层背面做好标记。用清洗剂洗干净、自来水冲洗、吹干后,依次置于丙酮、乙醇和异丙醇中超声30 min,吹干备用。
1.2.1.1.2 TiCl4的一次处理
将FTO玻璃两侧用胶带覆盖住各6 mm左右的宽度,然后置于40 mmol·L-1的TiCl4水溶液中,在70℃的温度下保温0.5 h,取出后先依次用蒸馏水和无水乙醇冲洗,然后放入马弗炉中以450℃的温度煅烧0.5 h。
1.2.1.1.3 TiO2光吸收层的胶体制备和滚涂
在1 mL蒸馏水中加入5滴乙酰丙酮,混合均匀后分批加入到3 g直径为25 nm的TiO2中,研磨均匀,研磨过程中加适量蒸馏水,这一过程持续25 min左右,然后加入5滴表面活性剂,仔细研磨均匀后再加入0.9 mL的醋酸氧化镁溶液,稍稍研磨均匀后装入安培瓶中,密封搅拌24 h。胶体搅拌好后,将FTO用胶带固定在台面上,FTO两侧被胶带覆盖的宽度在6 mm左右。在FTO上滴加适量胶体,迅速用玻璃棒滚涂均匀。待溶剂挥发后,将FTO置于马弗炉,以450℃的温度煅烧0.5 h。
1.2.1.1.4 TiO2光散射层胶体制备和滚涂
首先将乙基纤维素的乙醇溶液预先搅拌均匀。称取0.5 g颗粒直径为200 nm的TiO2粉末,倒入安培瓶中,再加入4.5 g溶解好的乙基纤维素溶液和0.1 mL醋酸氧化镁溶液,密封搅拌24 h。后续步骤与光吸收层的制作相同。
1.2.1.1.5 TiCl4的二次处理
与一次处理相同。
1.2.1.2DSSCs器件制作
光敏剂为染料的乙醇溶液。电解质的配方:0.05 mol·L-1I2、0.5 mol·L-1LiI、0.6 mol·L-11-甲基-3-己基咪唑碘、0.5 mol·L-14-叔丁基吡啶,溶剂采用3-甲氧基丙氰。将光阳极裁成1 cm×1.8 cm大小,100℃预热5 min后迅速泡入光敏剂溶液中,避光吸附12 h,取出后用乙醇冲洗,吹干,置于固定架上。光阳极上加入少量电解质后搭装铂电极,固定好后在模拟光源下进行测试。固定架底层用一块开孔面积为0.196 cm2的掩膜板来确定电池的活性面积。
1.2.2染料分子的合成
1.2.2.1化合物3a的合成
向100 mL的三口瓶中依次加入化合物1a(172 mg,0.6 mmol),化合物2(236 mg,0.5 mmol),Pd(PPh3)4(40 mg,0.03 mmol),K2CO3(2 mol·L-1,3 mL)和四氢呋喃(30 mL),抽真空,置换氩气,氩气保护下回流过夜。冷却至室温,加入100 mL水稀释,用二氯甲烷萃取,有机相饱和食盐水洗涤3次,再用无水MgSO4干燥。旋转蒸发除去溶剂,粗产品经硅胶柱层析,得到248 mg深红色固体化合物3a,产率85%。1H NMR(400 MHz,CDCl3):δ 10.14(s,1H),8.15 (d,J=7.6 Hz,1H),7.97(s,1H),7.63(q,J=7.4 Hz,4H),7.49(dd,J=19.9,10.8 Hz,4H),7.35~7.29(m,1H),7.22 (d,J=8.3 Hz,1H),3.23~3.08(m,2H),2.82(s,3H),2.79 (s,3H),2.57(s,3H),2.44(s,3H),1.80~1.67(m,2H),1.56~1.50(m,2H),1.42(dd,J=14.1,7.1 Hz,2H),0.95(t,J= 7.1 Hz,3H)。13C NMR(101 MHz,CDCl3):δ 186.26,159.89,155.33,148.70,141.34,140.42,138.94,137.68, 137.47,134.23,130.03,127.99,127.77,127.13,126.48, 125.87,124.00,123.68,123.03,121.94,120.39,120.32, 110.08,110.02,32.51,31.63,28.85,22.56,15.07,14.09, 14.07,13.01,12.65。
1.2.2.2化合物3b的合成
在100 mL的三口瓶中加入化合物1b和化合物2,用类似合成化合物3a的方法得到230 mg橙红色固体化合物3b,产率80%。1H NMR(400 MHz,CDCl3):δ 10.13(s,1H),8.20(dd,J=12.9,7.9 Hz,2H),7.65~7.55(m,4H),7.51~7.42(m,3H),7.36~7.30(m,1H),7.22(s,1H),7.11(d,J=7.7 Hz,1H),3.17~3.07(m,2H),2.80(s,3H),2.77(s,3H),2.52(s,3H),2.39(s,3H),1.78~1.62(m,2H),1.53~1.46(m,2H),1.39(dd,J= 13.8,6.8 Hz,2H),0.93(t,J=7.0 Hz,3H)。13C NMR (101 MHz,CDCl3):δ 186.22,159.30,155.52,148.92,141.39,141.00,140.21,139.24,139.18,137.43,134.06, 130.25,130.06,127.74,127.07,126.39,125.96,122.98, 122.08,120.48,120.42,120.33,111.27,109.96,32.47, 31.55,28.80,22.51,14.99,14.04,13.92,12.99,12.65。
1.2.2.3化合物3c的合成
在100 mL的三口瓶中加入化合物1c和化合物2,用类似合成化合物3a的方法得到250 mg橙红色固体化合物3c,产率86%。1H NMR(400 MHz,CDCl3):δ 10.15(s,1H),8.17(d,J=7.4 Hz,2H),7.70(d,J=7.2 Hz,2H),7.51(s,2H),7.46(d,J=6.5 Hz,4H),7.32 (s,2H),3.17(t,2H),2.83(s,3H),2.81(s,3H),2.63(s,3H),2.50(s,3H),1.74(s,2H),1.54(d,J=4.6 Hz,2H),1.43(d,J=6.5 Hz,2H),0.96(t,3H)。13C NMR(101 MHz, CDCl3):δ 186.34,158.42,149.58,140.67,137.39,131.73, 128.76,127.08,126.08,125.86,123.58,120.48,120.26, 119.64,116.62,109.78,109.73,32.53,31.66,28.92,22.57, 15.09,14.10,14.01,13.10,12.79。
1.2.2.4染料CB1的合成
向100 mL三口瓶中依次加入化合物3a(180 mg,0.3 mmol)、氰乙酸(50 mg,0.6 mmol)、乙腈(20 mL)、氯仿(20 mL)和哌啶(0.2 mL),在氩气保护下,80℃回流24 h。冷却至室温,反应液倒入50 mL二氯甲烷中稀释,水洗3次(3×50 mL),有机相无水MgSO4干燥。旋转蒸发除去溶剂,粗产物经硅胶柱层析(V石油醚∶V乙酸乙酯=10∶1),得到156 mg暗红色固体粉末,产率80%。1H NMR(600 MHz,DMSO):δ 8.30 (d,J=7.7 Hz,1H),8.23(s,1H),7.90(s,1H),7.68(dd,J= 20.7,7.1 Hz,4H),7.56(t,J=7.3 Hz,1H),7.45(t,J=8.0 Hz,2H),7.39(d,J=8.4 Hz,1H),7.34(d,J=8.3 Hz,1H),7.30(t,J=7.4 Hz,1H),3.11(t,2H),2.48(s,3H),2.47(s,3H),2.42(s,3H),2.41(s,3H),1.64(dd,J=14.8,7.7 Hz,2H),1.48(dd,J=14.6,7.5 Hz,2H),1.40~1.37(m,2H),0.90(dd,J=7.2,3.4 Hz,3H)。13C NMR(151 MHz,DMSO):δ163.65,155.53,150.83,148.31,141.58,140.94, 140.69,139.87,139.12,138.45,137.62,137.39,137.19, 135.60,132.20,132.00,130.99,130.61,129.12,128.85, 128.28,127.18,126.07,124.62,123.50,123.10,122.79, 121.35,120.70,120.26,119.32,109.92,109.45,32.34, 30.46,29.02,22.31,19.11,15.37,14.81,14.01,13.17。MALDI-TOF-MS,m/z:Calcd.for C39H35BF2N4O2[M]+:654.300,Found:654.307。
1.2.2.5染料CB2的合成
用类似于合成染料CB1的方法得到140 mg深红色固体粉末CB2,产率75%。1H NMR(600 MHz,DMSO):δ 8.34(d,J=7.8 Hz,1H),8.29(d,J=7.2 Hz,1H),7.89(s,1H),7.68~7.64(m,4H),7.53(d,J=6.1 Hz,1H),7.48~7.44(m,1H),7.40(d,J=7.9 Hz,1H),7.34~7.30(m,1H),7.21(s,2H),3.05(d,J=0.8 Hz,2H),2.46 (s,3H),2.41(s,3H),2.40(s,3H),2.34(s,3H),1.70~1.66 (m,2H),1.47~1.43(m,2H),1.34(d,J=6.7 Hz,2H),0.87 (t,J=6.7 Hz,3H)。13C NMR(151 MHz,DMSO):δ 163.55, 155.04,151.24,148.58,141.01,140.61,139.11,137.74, 137.10,135.45,132.17,131.37,131.00,130.57,128.20, 127.05,126.04,123.33,122.88,122.55,121.14,120.78, 119.02,111.47,110.15,32.30,31.37,28.56,22.25,15.41, 14.71,14.35,13.86,13.07。MALDI-TOF-MS,m/z:Calcd. for C39H35BF2N4O2([M]+):654.300,Found:654.305。
1.2.2.6染料CB3的合成
用类似于合成染料CB1的方法得到153 mg紫红色固体粉末CB3,产率78%。1H NMR(600 MHz,DMSO):δ 8.21(d,J=7.7 Hz,2H),8.12(s,1H),7.67~7.69(d,J=7.7 Hz,2H),7.62(d,J=7.7 Hz,2H),7.47(d,J=9.8 Hz,2H),7.36(d,J=7.2 Hz,2H),7.19~7.24(t,J= 7.3 Hz,2H),3.12(s,2H),2.47(s,3H),2.46(s,3H),2.43 (s,3H),2.41(s,3H),1.79(m,2H),1.65(m,2H),1.49(m,2H),0.90(t,J=7.0 Hz,3H)。13C NMR(151 MHz,DMSO):δ174.83,172.89,140.75,140.49,137.63,133.49, 132.47,131.41,131.32,130.12,126.81,126.68,126.48, 123.29,123.07,120.93,120.32,110.29,110.23,32.94, 31.75,29.49,22.56,15.43,14.49,14.42,13.64,13.22。MALDI-TOF-MS,m/z:Calcd.for C39H35BF2N4O2([M]+):654.300,Found:654.318。
2 结果与讨论
2.1染料的合成
染料CB1~CB3的合成路线如图式2所示。化合物1a~1c和2均按照文献方法进行制备。1a~1c与2在碳酸钾和四(三苯基膦)钯的作用下,通过经典的Suzuki偶联反应可以高效合成重要的中间体3a~3c,其收率均在80%以上。化合物3a~3c与2-氰基乙酸在吡啶的作用下通过经典的Knoevenagel反应得到最终的染料CB1~CB3。从原料到最终的染料,总的反应收率在60%以上,且分离纯化较为容易。染料CB1~CB3的结构通过1H,13C NMR以及MALDI-TOF-MS进行了确证。
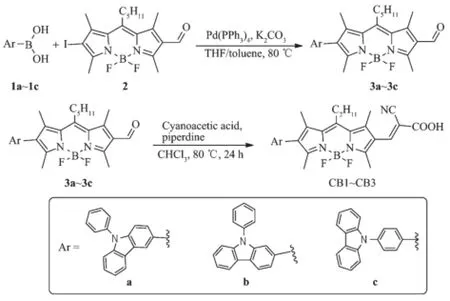
图式2 BODIPY染料敏化剂CB1~CB3的合成路线Scheme 2 Synthetic routes of BODIPY dye sensitizers (CB1~CB3)
2.2染料的光物理性质
染料CB1~CB3在氯仿溶液(c=10-5mol·L-1)中的紫外-可见吸收光谱(UV-Vis)如图1所示,其对应的光谱数据列于表1。从图1(上)中可以看到染料在CB1~CB3在250~400 nm和420~600 nm之间有2组吸收峰。其中,能量较高的一组是由N-苯基咔唑单元的π-π*迁移引起的吸收峰;能量较低的一组则是由于电子给体单元与电子受体单元之间的分子内的电子迁移(ICT)所引起的迁移吸收。如图1(a)所示BODIPY母核的最大吸收峰位于498 nm(BDP core),而CB1~CB3在BODIPY的2,6-位分别引入了N-苯基咔唑单元和氰乙酸受体单元以后,吸收光谱产生了30 nm左右的红移,这说明分子的有效共轭长度增加,分子内的电子迁移增强。与CB1和CB3相比,CB2的吸收光谱更宽,吸收强度更大,这说明了N-苯基咔唑的2-位与BODIPY母核相连较3-位和苯环4-位与BODIPY母核相连产生了更强的共轭效应,更有利于分子的光谱吸收和分子内电荷迁移。除此之外,3种染料都展现出了良好的集光性能,其在各自最大吸收峰处的摩尔消光系数分别高达5.6×104、7.4×104和5.4×104L·mol-1·cm-1。CB2比CB1和CB3的摩尔消光系数更高可能是其较为优良的结构特点引起的。以上结果表明,该类BODIPY染料分子具有良好的光子捕获能力,具备了作为染料敏化剂所必需的光谱条件。
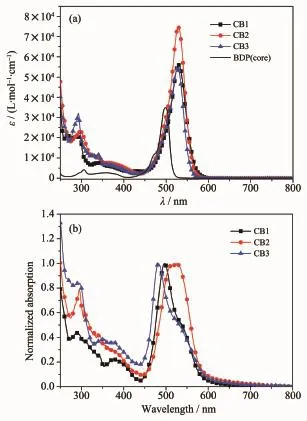
图1染料CB1~CB3在氯仿中(a)的紫外-可见光吸收光谱和吸附在TiO2薄膜上(b)的归一化紫外-可见光吸收光谱Fig.1 UV-Vis absorption spectra of dyes CB1~CB3 in CHCl3(a)and normalized absorption spectra of dyes CB1~CB3 adsorbed on TiO2films(b)
通常来讲,当染料吸附到纳米晶TiO2表面时,染料的去质子化效应和聚集效应会对其吸收光谱产生一定的影响。去质子化效应和H-聚集一般会产生光谱吸收的蓝移,而J-聚集则会产生光谱吸收的红移[31]。如图1(b)所示,染料CB1~CB3吸附于TiO2膜上(3 μm)的最大吸收峰分别位于505、524和497 nm。与其在溶液中的光谱吸收相比,分别出现了24、5和29 nm的蓝移,这说明染料CB1~CB3在纳米晶TiO2表面发生去质子化效应和H-聚集。相比于CB1和CB3,CB2的蓝移现象较为微弱,这预示着其较低程度的聚集和较高的光伏性能。
染料CB1~CB3在氯仿溶液(c=1×10-7mol·L-1)中的荧光光谱如图2所示。3种染料在不同极性溶剂中的发射光谱均未发生明显的变化,这说明其在溶液中的发射光谱不是由于溶剂效应和聚集效应引起的发光,而是染料CB1~CB3的本征迁移发光。采用2个最大吸收波长进行激发(例如CB2,其激发波长分别为298和529 nm),得到相同的荧光发射波谱,说明了激子从受体单元到给体单元之间的能量转移。这种现象表明3种染料中的给电子单元、桥联基团和吸电子单元所吸收的光子均可以有效的用于光电转换中。CB1~CB3的荧光发射峰分别位于573、565和570 nm。相较于CB2,CB1和CB3的发射光谱表现出8~5 nm的红移,这说明N-苯基咔唑单元不同的位置与BODIPY相连对其光物理性质有一定的影响。

图2染料CB1~CB3在氯仿(c=1×10-7mol·L-1)中的归一化荧光光谱图Fig.2 Normalized fluorescence spectra of dyes CB1~CB3 in CHCl3(c=1×10-7mol·L-1)
2.3染料的电化学性质研究
为了研究BODIPY染料CB1~CB3激发态电子能否顺利注入到TiO2的导带中,我们用循环伏安法测量了3种染料在二氯甲烷溶液中的氧化/还原行为,结果见图3和表1。循环伏安曲线测试是利用三电极体系测定的,其中工作电极是金电极,对电极为铂丝电极,参比电极为Ag/AgCl电极,支持电解质为四丁基六氟磷酸铵(0.1 mol·L-1)的二氯甲烷溶液,扫描速度为100 mV·s-1,参比电极以二茂铁为内标进行标定,其相对于标准氢电极电势为0.63 V。染料的HOMO能级与LUMO能级之间的能隙E0,0可以通过紫外吸收光谱阈值波长计算得到。半波氧化电位(Eox)通过测试得到,激发态还原电位E*ox=Eox-E0,0。结果表明染料分子的基态氧化电位(Eoxvs NHE)均比I3-/I-电对的氧化/还原电位(0.42 V vs NHE)更正,即失去电子的氧化态染料分子能够有效被I-还原;染料分子的激发态还原电位均比二氧化钛的导带的能级(-0.5 V vs NHE)更负,才能使激发态的电子有效注入到半导体的导带,这样基于染料电池的电子循环得到了充分的利用,因而这些BODIPY染料可以作为二氧化钛电极的敏化剂。
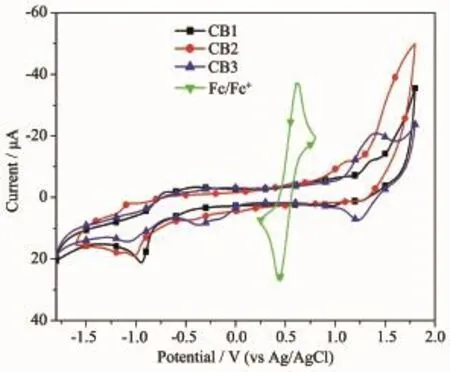
图3染料CB1~CB3在CH2Cl2溶液中的循环伏安曲线Fig.3 Cyclic voltammograms curves of dyes CB1~CB3measured in CH2Cl2solution
2.4染料分子的理论计算
为了深入理解染料的结构和光电性能之间的关系,我们利用量子化学密度泛函理论计算的方法对染料CB1~CB3的结构和电子云分布情况进行了模拟研究,所有的染料分子的初始结构均由GaussView5.0构建,采用Gaussian09程序,选用的方法与基组分别为B3LYP和6-31G(d,p)。CB1~3优化的结构和LUMO与HOMO能级电子分布列于图4和图5。从图4中所示的CB1~3的基态几何结构中可以看出:CB1中咔唑平面与BODIPY平面之间的二面角为46.8°,CB2中咔唑平面与BODIPY平面之间的二面角为43.6°,而CB3中咔唑平面与BODIPY平面之间的二面角则为57.1°,三者中CB2的二面角最小,说明CB2的整个分子更接近于共平面结构,这有利于分子内的电子迁移(ICT)和增加分子的有效共轭长度,从而产生吸收光谱的拓宽和红移,这与前文提及的CB2具有更为宽阔和强烈的光谱吸收特征吻合。这也进一步预示了CB2较为优异的光伏性能。
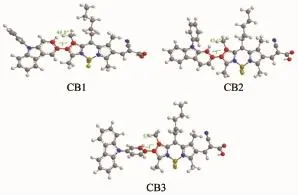
图4染料CB1~CB3的最优几何构型Fig.4 Optimized structures of dyes CB1~CB3
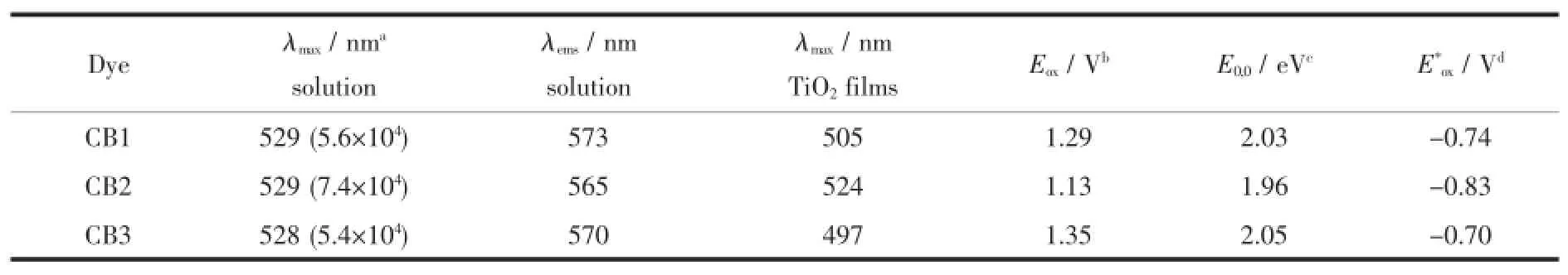
表1染料CB1~3的光物理和电化学性能数据Table 1 Photophysical and electrochem ical properties data of dyes CB1~CB3
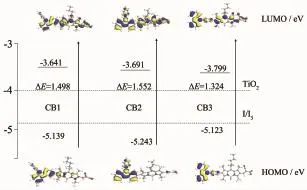
图5染料CB1~CB3的HOMO和LUMO量化计算能级Fig.5 Calculated HOMO and LUMO levels for dyes CB1~CB3
从优化的基态和激发态前线分子轨道图和电子分布图可以看出:染料CB1~CB3的LUMOHOMO能级结构分别为3.641/5.139 eV,3.691/5.243 eV和3.799/5.123 eV。基态时电子云主要分布于作为供电基团的N-苯基咔唑上;激发态时电子云大部分布居于与其相连的π桥和受体基团上。这说明光激发的电子能有效的从给电子单元分离到电子受体单元,这对激发态的电子有效的注入TiO2的导带至关重要。然而,我们也可以清楚的看到,在LUMO能级时,给电子基团上也有少量电子云分布,这主要由分子的几何结构引起的。因为如图4所示,给体单元与受体单元之间较大的二面角抑制了分子内电荷迁移,使得光电子的分离不完全。相对于CB1和CB3,CB2在LUMO能级时,电子受体单元的电子云密度更大,这也预示着CB2较好的电荷分离性能。从图5所显示的染料的LUMO能级数据可以看出,所有染料的LUMO能级都高于TiO2的导带能级,说明所有激发态的染料分子都能够有效地将电子注入到TiO2导带中;其HOMO能级均低于I/I3-的氧化/还原能级,说明所有处于氧化态的染料分子能够及时的在电解液中被还原至基态,从而进行循环再生利用。这些分子能级和电子分布数据表明设计合成的所有染料从理论上均符合染料敏化太阳能电池纯有机染料敏化剂要求。
2.5染料分子的光伏性能
在AM1.5(100 mW·cm-2)的模拟太阳光下,基于CB1~CB3敏化的太阳能电池的电流-电压(J-V)曲线如图6所示,相应的短路电流(Jsc)、开路电压(Voc)、填充因子(FF)、光电转换效率(PCE)等电池参数列于表2。CB3敏化的太阳能电池器件的能量转换效率为0.65%,其Jsc为1.68 mA·cm-2,Voc为530 mV,FF值为0.73。CB1的Voc值与CB3一样,但是相比CB3,CB1的Jsc值有明显的提高,达到2.61 mA·cm-2,FF值也略有提高,为0.76。使得CB1的电池参数较高的原因可能是N-苯基咔唑的3-位与BODIPY单元相连,给电子中心更靠近受体单元,因而更有利于光电子的分子内迁移;同时,由于在CB1中,N-苯基咔唑给电子单元与桥联基团BODIPY单元之间的二面角更小,有利于共轭π电子的离域。而给电子中心更靠近桥联基团且两者的二面角更小的CB2则展现了更好的光谱吸收性能和光伏性能。基于CB2的太阳能电池的Jsc达到3.71 mA·cm-2,Voc为550 mV,填充因子FF为0.73,总光电转换效率为1.49%。这充分说明,染料的电子给体和受体之间的距离和分子内电子传输、分离对其光伏性能有较大的影响。相比于CB1和CB3,基于CB2的DSSC拥有更高的Voc值,可能是由于CB2吸附的TiO2半导体具有较高的导带能级.这与理论计算的结果一致。
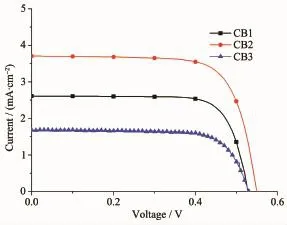
图6染料CB1~CB3的电流-电压(J-V)曲线Fig.6 Current-potential(J-V)curve for dyes CB1~CB3

表2染料CB1~CB3敏化DSSCs的性能参数Table 2 Parameters of DSSCs based on CB1~CB3
3 结论
本文合成了3种基于N-苯基咔唑和BODIPY的D-π-A型光敏染料(CB1~CB3)。光物理、电化学性质和光伏性能研究及理论计算结果显示:3种染料具有宽的光谱响应范围和高摩尔消光系数,有较为适合的能级结构,满足染料光敏剂的要求。结构-性能关系结果表明:N-苯基咔唑的咔唑环与BODIPY桥相连的分子(CB1,CB2)表现出更优良的光电性质;且咔唑环上2-位取代的分子(CB2)比3-位取代的分子(CB3)具有更优良的光谱吸收、能级结构及光伏性能。基于CB2的染料敏化太阳能电池的总光电转换效率达到1.49%,但是光电流仍然较低。因此,提升BODIPY类染料敏化电池的光电流仍是今后研究的重点。
[1]O′Regan B,Grätzel M.Nature,1991,353:737-739
[2]Ahmad S,Guillen E,Kavan L,et al.Energy Environ.Sci., 2013,6:3439-3466
[3]Yousuke O,Yutaka H.Eur.J.Org.Chem.,2009,18:2903-2934
[4]Han L Y,Islam A,Chen H,et al.Energy.Environ.Sci., 2012,5:6057-6060
[5]Numata Y,Singh S P,Islam A,et al.Adv.Funct.Mater., 2013,23:1817-1823
[6]Mishra M,Fischer M,Buerle P.Angew.Chem.Int.Ed., 2009,48:2474-2499
[7]Mao J Y,He N N,Ning Z J,et al.Angew.Chem.Int.Ed., 2012,51:9873-9876
[8]Yang J B,Ganesan P,Teuscher J,et al.J.Am.Chem.Soc., 2014,136:5722-5730
[9]Wang Z S,Cui Y,Dan-oh Y.J.Phys.Chem.C,2007,111: 7224-7230
[10]Chen Y S,Li C,Zeng Z H,et al.J.Mater.Chem.,2005,15: 1654-1661
[11]Li C,Yum J,Moon S,et al.ChemSusChem.,2008,1:615-618
[12]Yella A,Lee H W,Tsao H N,et al.Science,2011,334:629 -634
[13]Wang S B,Guo J C,He L,et al.Synth.Met.,2013,168:1-8
[14]Jian B W,Xia Q F,Xu P,et al.Chem.Asian J.,2012,7: 696-700
[15]Zhang S,Wang Y,Cui K,et al.Adv.Mater.,2007,19:1138-1141
[16]Liang M,Xu W,Cai F,et al.J.Phys.Chem.C,2007,111: 4465-4472
[17]Loudet A,Burgess K.Chem.Rev.,2007,107:4891-4932
[18]Ziessel R,Ulrich G,Harriman A.New J.Chem.,2007,31: 496-501
[19]Lefebvre J F,Sun X Z,Calladine J A,et al.Chem.Commun., 2014,50:5258-5260
[20]Kolemen S,Bozdemir O A,Cakmak Y,et al.Chem.Sci., 2011,2:949-954
[21]Warnan J,Buchet F,Pellegrin Y,et al.Org.Lett.,2011,13: 3944-3947
[22]Mao M,Zhang X L,Cao L,et al.Dyes Pigm.,2015,117:28-36
[23]Bura T,Leclerc N,Fall S.J.Am.Chem.Soc.,2012,134: 17404-17407
[24]Zhao H B,Wang B Y,Liao J X,et al.Tetrahedron Lett., 2013,54:6019-6022
[25]Mao M,Zhang X L,Fang X Q,et al.Org.Electron.,2014, 15:2079-2090
[26]Koumura N,Hara K.Heterocycles,2013,87:275-301
[27]Venkateswararao A,Thomas K R Justin,Lee C P,et al. ACS Appl.Mater.Interfaces,2014,6:2528-2539
[28]Ho C L,Wong W Y,Yao B,et al.J.Organomet.Chem., 2009,694:2735-2749
[29]Li J Y,Wang R J,Yang R X,et al.J.Mater.Chem.C, 2013,26:4171-4179
[30]Wang K Y,Chen C,Liu J F,et al.Org.Biomol.Chem., 2012,10:6693-6704
[31]Shen P,Liu X P,Jiang S H,et al.Org.Electron.,2011,12: 1992-2002
Synthesis and Properties of 2,6-Modified BODIPY Sensitizers for Dye-Sensitized Solar Cells
ZHOU Wei-Nan1,2ZHAO Hong-Bin*,2CAI Zhuo-Di3LIAO Jun-Xu*,2,3PENG Fei1,2YAN Wen-Jie2YANG Nian-Fa*,1
(1College of Chemistry,Xiangtan University,Xiangtan,Hunan 411105,China)
(2School of Chemistry and Environmental Engineering,Dongguan University of Technology,Dongguan,Guangdong 523808,China)
(3Chemical Industry and Engineering Society of Dongguan,Dongguan,Guangdong 523808,China)
Three novel D-π-A type organic dye sensitizers based on BODIPY derivatives featured with BODIPY as π-conjugate bridge,N-phenylcarbazole linked BODIPY unit with different active positions as electron donor and cyanoacrylic acid as electron acceptor were designed and successfully synthesized.The structures of these synthetic dyes CB1~CB3 were characterized by1H,13C NMR and MALDI-TOF-MS.The photophysical properties, electrochemical behaviors and photovoltaic performances of these dyes were measured and the relationships between the structure and property were further studied by density functional theory calculations.The absorption spectral of CB1~CB3 ranged from 420 nm to 600 nm.Their LUMO and HOMO levels were located on about 3.7 eV and 5.2 eV respectively.CB2 exhibited more excellent photovoltaic performance than CB1 and CB3 due to the more planar structure and more suitable conjugation length,which resulting in broad absorption and betterintramolecular electron transfer property.Under AM 1.5(100 mW·cm-2)irradiation,CB2 sensitized solar cell give 1.49%total photoelectric conversion efficiency with open circuit voltage(Voc)550 mV,short-circuit current density(Jsc)3.71mA·cm-2and fill factor(FF)0.73.
dye-sensitized solar cells;organic dyes;photovoltaic performance;BODIPY
O626.1
A
1001-4861(2016)01-0081-08
10.11862/CJIC.2016.021
2015-07-20。收修改稿日期:2015-10-30。
国家自然科学基金(No.21342003),广东省自然科学基金(No.2014A030313630),广东省教育厅高层次人才培养项目(No.粤财教(2013)246号),广东省高校科技创新项目(No.cxzd1148),东莞市高等院校科研机构科技计划项目(No.2012108101005)资助。
*通信联系人。
E-mail:zhaohbhanlf@163.com
猜你喜欢
辽宁化工(2022年8期)2022-08-27
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年8期)2021-12-21
石油炼制与化工(2021年12期)2021-12-14
世界科学技术-中医药现代化(2021年7期)2021-11-04
河南科学(2020年6期)2020-07-27
农药科学与管理(2019年8期)2019-11-23
原子与分子物理学报(2019年5期)2019-04-28
陶瓷学报(2019年5期)2019-01-12
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
