
基于多样本RNA-Seq数据的表达水平估计方法*
2016-11-30 09:43刘学军陈松灿
计算机与生活 2016年2期
张 礼,刘学军,陈松灿
南京航空航天大学 计算机科学与技术学院,南京 210016
基于多样本RNA-Seq数据的表达水平估计方法*
张礼+,刘学军,陈松灿
南京航空航天大学 计算机科学与技术学院,南京 210016
ZHANG Li,LIU Xuejun,CHEN Songcan.Novel method to estimate expression level based on multi-sample RNA-Seq data.Journal of Frontiers of Computer Science and Technology,2016,10(2):210-219.
随着下一代高通量DNA测序的快速发展,RNA-Seq测序已成为转录组学分析的标准技术。在处理多样本RNA-Seq数据时,现有表达水平估计方法通常基于单个样本逐个处理,忽略了基因读段分布在样本间高度相似的特点。因此,提出了一个基于多样本RNA-Seq数据的表达水平估计方法,称为MRSeq。其关键是通过建立偏差曲线估计模型获得基因读段分布在样本之间的共享特征,通过偏差权重将共享特征嵌入到模型中,用来修正读段数据,同时通过增加稀疏约束来表现基因和异构体表达水平之间的稀疏性。进而将该模型应用到多个真实数据集进行评测,与目前主流方法的比较结果表明:MRSeq不仅能得到准确的基因和异构体表达水平,同时也获得了更有意义的生物解释。
RNA-Seq;多样本;偏差曲线;稀疏;基因和异构体表达水平
1 引言
近几年来,下一代高通量测序技术得到快速发展,RNA-Seq(RNA sequencing)测序技术被广泛应用到转录组学的研究上[1-2]。与传统的基因芯片技术相比,RNA-Seq可在全基因组范围内进行测序,具有高通量,高灵敏度,可重复性好,样本需求低等特点,具有广泛的应用场景,比如估计基因或异构体表达水平,寻找差异表达基因或异构体,发现未知的异构体等,正快速成为研究转录组学的基本实验手段[3-4]。
当从RNA-Seq测序实验获得海量读段数据后,计算基因和异构体的表达水平是研究转录组学中最为基础的实验目的[5-6]。rSeq(RNA-Seq analyzer)方法假设基因的每个外显子上的读段数目服从泊松分布,其参数是基因所包含的每个异构体表达水平的线性加权和。rSeq方法解决了因选择性剪接而造成的读段多源映射问题,可同时估计基因和异构体表达水平[7]。但rSeq方法是基于基因读段分布是均匀的假设,而实际情况中,读段分布具有明显的非均匀特征,该特征通常是由两类原因造成的:其一是技术性偏差,在RNA-Seq测序技术过程中,研究人员经常使用不同的方法进行RNA提取、大小选择、片段化、转换为cDNA(complementary DNA)、扩增和最后测序,这导致同一个样本的多次测序结果存在偏差[8]。其二是生物性偏差,如制备cDNA文库时,RNA序列对反转录所采用的随机引物具有一定偏好,在PCR扩增阶段,cDNA片段倾向GC碱基含量高的片段、ploy(A)端和低复杂度的序列[9-10]。技术上和生物上的偏差导致读段分布具有非均匀分布的特征。NURD(non-uniform read distribution)方法改进了rSeq模型,通过加入预先计算的基于基因的全局偏差权重和基于异构体的局部偏差权重,来模拟读段分布的非均匀分布特征[11-12]。POM(Poisson mixed-effects model)方法采用基于碱基层面的泊松分布,构建一个统计模型来获得序列中每个碱基的位置偏差和碱基之间的相互作用[13]。GP(generalized Poisson)和WemIQ方法采用广义泊松分布,通过增加额外参数来解决因读段非均匀分布而造成的过散布问题,相比传统的泊松分布能更好地拟合数据[14-15]。Cufflinks方法把偏差特征分为位置偏差和序列偏差,通过分别建模来计算不同偏差的影响,进一步量化偏差的构成和更准确地模拟读段采样的随机特性[16-17]。RSEM(RNASeq by expectation-maximization)和BitSeq(Bayesian inference of transcripts from sequencing Data)方法都采用了产生式概率图模型,模拟读段产生过程中的经验分布,以及考虑额外的数据匹配信息,如匹配质量、映射方向等[18-20]。但两者采用不同的方式来考虑读段分布的非均匀特征。RSEM利用读段起始位置的经验分布来表示非均匀特征,而BitSeq采用了与Cufflinks同样的偏差模型[20]。采用不同的偏差计算方式来模拟读段非均匀分布,上述方法都能提高基因和异构体表达水平的准确程度。
在RNA-Seq测序实验中,为了避免实验中的技术性误差,同一个测序样本会进行多次技术性重复实验,从而获得多样本RNA-Seq数据。但是上述方法通常是逐个处理每个样本,导致数据中的技术性偏差难以消除,同时不可避免地丢失了样本之间的相关信息。如图1所示(数据来自3.1节中的小鼠数据集),老鼠基因APOE在其大脑组织的两个样本中,其读段分布具有高度的相似性,其相关系数高达0.96。说明同一个基因的读段分布模式在不同样本中是具有很高相关性的。在分析转录组的其他任务中,如寻找差异基因和异构体表达水平中,BDSeq方法考虑了多样本RNA-Seq数据之间关联获得更为精确的结果[21];寻找新的异构体方法中,MITIE(mixed integertranscript identification)方法发现精度随着样本数目增多而提高[22]。这些方法证明了考虑多样本数据之间的相关性能帮助提高精度。而在估计表达水平中,很少有方法结合多个样本数据来计算基因和异构体表达水平。
在多样本数据中,同一个基因中的某个异构体,在不同技术性重复样本中其表达水平应该是相近的。但由于技术性偏差以及数据噪声,导致有些异构体的表达水平出现偏差。其次同一个基因虽包含多个异构体,但基因发生表达时通常由少数异构体所体现出来,具有稀疏性的特点[23]。比如人类基因ENSG000000040597包含6个异构体,表1是采用Cufflinks方法计算出的6个异构体在不同大脑样本中的表达水平。从表1中可看出,在不同样本中基因ENSG000000040597通常有4或5个异构体发生表达,但起主要作用的是由ENST00000000233和ENST00000489671两个异构体表现出来的,ENST00000415666和ENST00000463733两个异构体是不确定的低表达,ENST00000459680异构体不表达,而ENST00000467281在不同样本的表达水平差异很大且表达值很低,受到偏差和数据噪声的影响。因此,逐个处理单个样本的方法不能很好地表现生物体本身所蕴含的生物特性。
基于上述问题,本文提出了一个基于多样本RNA-Seq数据的回归模型来估计基因和异构体表达水平,MRSeq(multi-sample RNA-Seq model)。考虑基因在多样本中的读段分布具有高度相似性,设计了一个联合多样本数据的偏差模型,能更为准确地表现基因读段分布在样本之间的共享特征。同时增加了L2/L1稀疏约束,用来消除技术性偏差给基因和异构体在不同样本中带来的差异,且保留基因和异构体表达水平之间的稀疏性质,更好地表现其生物特性,获得更有意义的生物解释。通过老鼠和人类的多个真实数据集来验证MRSeq的有效性。
2 方法
2.1MRSeq模型
因选择性剪接在真核生物中是普遍存在的,外显子片段的选取导致基因通常包含多个异构体。假设基因g包含K个异构体,其异构体与基因之间的关系可使用数学模型二元矩阵A=(aik)M×K准确表示,其中M表示基因g包含M个外显子,其长度分别为l1,l2…,lm。每个异构体对应矩阵A中的一列,其中(aik)=1表示第k个异构体包含第i个外显子,反之则为0。
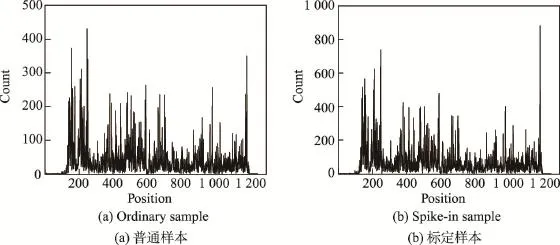
Fig.1 Read distribution ofAPOE gene in two mouse brain samples图1 老鼠基因APOE在两个大脑组织样本中的读段分布
假设RNA-Seq测序实验获得N个样本,对于基因g,yij表示第i个外显子在第j个样本中观测到的读段数目。根据实验原理,yij等同于基因g在第j个样本中所包含异构体中第i个外显子上的读段之和:

其中,xkj表示第j个样本中第k个异构体上期望的读段数目。当获得xkj后,便可计算异构体的FPKM(fragments per kilobase of transcript per million mapped reads)值,Wj是第j个样本中的总读段数目[15]。
式(1)模型是假设读段在均匀分布的情况下,但在实际数据中,读段具有明显的非均匀分布特征。假设bi表示第i个外显子的偏差权重。因为基因的读段分布模式在不同样本中是高度相似的,所以假设bi在不同样本中是共享的。因此式(1)可改写成:

对于基因g,其K个异构体在N个样本中表达水平X可通过回归模型估计出来,其公式如下:
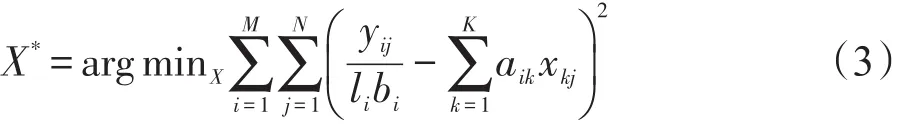
对于所有异构体表达水平xkj≥0。
同一个基因中相同异构体在不同的技术性重复样本中,其表达水平应该是接近的,但在实际数据中,因为偏差和数据噪声的影响,造成表达水平在不同样本中具有差异性,特别是低表达水平的异构体和基因。其次,同一个基因虽包含多个异构体,但在基因表达时通常由少数异构体体现出来[23],基因和异构体表达水平之间是具有稀疏性的。为了融入数据中的先验信息,MRSeq方法增加了L2/L1稀疏约束,式(3)可写成如下:
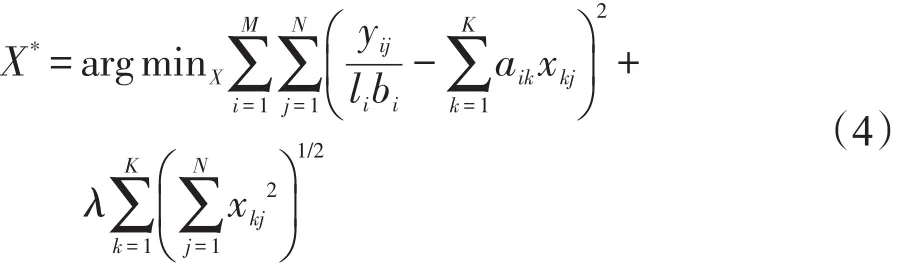
为了方便计算,式(4)可简化成矩阵形式:
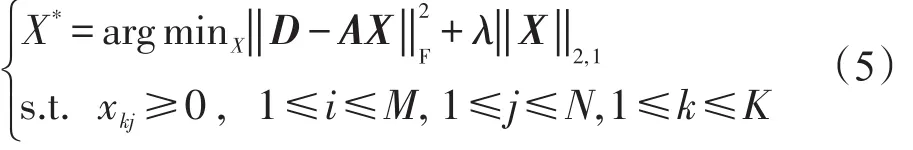
通过优化MRSeq方法的目标函数(5),即可获得异构体在不同样本中的表达水平,其基因表达水平等于对应异构体表达水平之和。当λ→+∞,X*=0表示异构体在不同样本中都没有表达。随着λ的减小,矩阵X*的某些行不再为0,表示对应的异构体发生表达。本文的所有实验都选择λ=1。选择一个最优的λ是至关重要的,在3.6节中将讨论λ的选择问题。
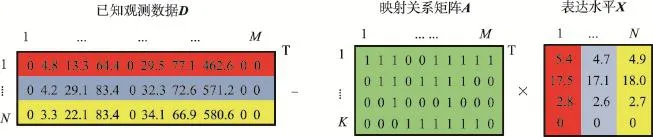
Fig.2 Core optimization problem of MRSeq图2 MRSeq方法的核心优化问题
2.2多样本数据的偏差曲线估计
在多样本数据中,基因的读段分布在样本之间具有高度相似性。本文提出一种多样本数据的偏差估计模型来表示基因读段分布在样本之间的共享特征。选择只包含单个异构体的基因,因为多异构体的基因其读段分布受到基因结构的影响,不能正确反映基因中读段的分布趋势。由于低表达水平基因的不确定性,本文排除读段数目小于50的基因。为了避免读段分布的局部影响,把基因的长度划为S个区间(通常选择20),统计每个区间内的读段数目,采用均值为一的方法来归一化区间的读段数目。采用多项式回归模型来拟合归一化后每个区间的读段数目,估计出光滑的曲线来描述多样本数据的偏差曲线。具体算法步骤如图3中步骤1所示。
2.3模型实现
在获得多样本读段匹配数据,MRSeq方法的实现分为两个步骤:数据预处理和表达水平计算。MRSeq方法预处理过程主要包括统计基因外显子的读段数目以及计算基因的偏差权重。在表达水平计算步骤上,因为目标函数是带L2/L1稀疏约束的凸优化问题,采用SPAMS(sparse modeling software)优化工具箱来求解[24]。SPAMS是一个为解决各种稀疏估计问题,提供多种语言接口和跨多平台的开源优化工具箱。MRSeq方法的详细流程如算法1所示。
算法1 MRSeq


为了方便用户理解和使用MRSeq方法,本文提供了一个系统的RNA-Seq实验数据分析通道,如图4所示。当RNA-Seq测序实验获得N个读段数据样本后,使用最流行的匹配软件(Bowtie(v.2.2.3))来匹配读段到已知的参考转路本序列[25]。匹配成功的读段数据作为MRSeq方法的输入,而MRSeq方法输出的基因和异构体表达水平可提供给后续分析使用,比如寻找差异基因或异构体表达,基因网络分析等。
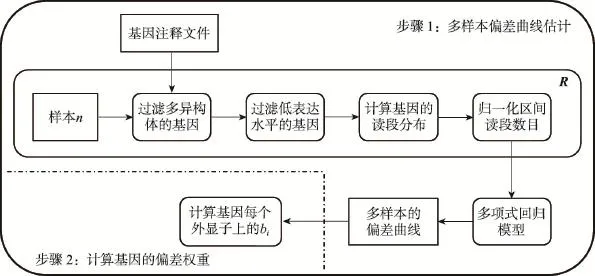
Fig.3 Workflow of bias weight calculation图3 计算基因的偏差权重的流程图
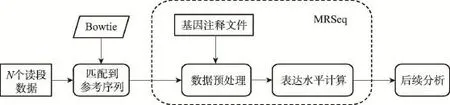
Fig.4 Pipeline of RNA-Seq data analysis图4 RNA-Seq数据分析通道的流程图
3 实验结果分析
本文选择3个主流方法Cufflinks(v.2.2.1)、RSEM(v.1.2.19)和NURD(v.1.1.1)与MRSeq方法在3个数据集上进行比较,用来验证基因和异构体表达水平。
3.1实验数据集
MRSeq方法可同时估计基因和异构体表达水平,本文选择3个真实RNA-Seq数据集来评估分法性能。数据集都来自Illumina/solexa测序平台,基因注释信息都来自UCSCGenome Browser。
小鼠数据集包含大脑、肝脏和骨骼肌3个组织,其中每个组织分别包含两个技术性重复实验样本。使用RefSeq数据库的基因注释信息(GRCm38/mm10),总共包含33 608个异构体,主要用来验证样本之间的异构体表达水平[26]。
人类大脑数据集来自美国药品监管局(FDA)联合全球多所高校研究机构进行的“生物芯片质量控制(MAQC)”项目。MAQC数据集是评估计算基因表达水平模型的标准数据集,被最为广泛地使用。此数据集包含单末端(single-end,SE)和双末端(pairedend,PE)两种类型的读段数据。单末端读段数据集包含两个条件,正常大脑组织(HBR)和病变大脑组织(UHR),每个条件下包括7次测序通道,相当于7次技术性重复实验样本。双末端读段数据集只有正常大脑组织(HBR),包括3次测序通道。使用Ensembl数据库的基因注释信息(GRCh37/hg19)。MAQC项目提供了1 000个qRT-PCR(quantitative real-time PCR)验证基因,根据与Ensembl注释库的对应匹配,最终获得833个qRT-PCR验证基因。这些基因的qRT-PCR值被认为是基因的真实表达水平,可被用来评估模型计算基因表达水平的准确程度[27]。
人类乳腺癌数据集有两个条件,乳腺正常细胞(HME)和乳腺癌细胞(MCF-7),分别包括4个和7个测序通道。文献[28]提供了5个基因中8个异构体的qRT-PCR验证值,可用来评估模型计算异构体表达水平的准确程度。根据文献选择UCSC注释库的基因注释信息(GRCh36/hg18)[28]。
3.2多样本数据的偏差曲线
为了验证多样本偏差曲线估计模型,选择MAQC在HBR条件下的单末端数据集,采用图3中步骤1的计算流程,把过滤后的基因分成20个区间进行读段数目统计和归一化,选择多项式回归模型来拟合7个样本获得的数据,最终得到如图5(a)所示的偏差曲线。图5(a)中的偏差曲线显示在基因的读段分布是具有明显非均匀分布特征的。基因的3′端和5′端是最容易受到偏差影响的[12,17],正好对应着偏差曲线中的两端。
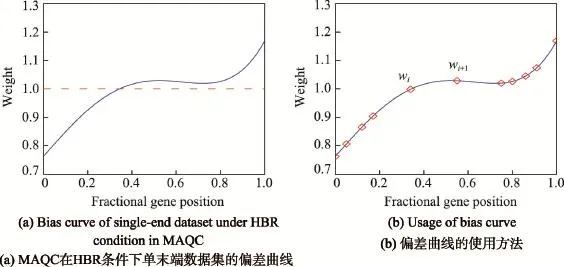
Fig.5 Bias curves of multi-samples图5 多样本偏差曲线
3.3多样本数据之间的表达水平验证
在多样本数据中,同一个基因中相同异构体在不同技术性重复样本中其表达水平应该是相近的,且当基因发生表达时通常是由其包含的少数异构体所表现出来的,此处主要验证异构体在样本之间的表达水平。表1给出了Cufflinks方法估计的异构体表达水平,基因ENSG000000040597通常有4或5个异构体发生表达,且ENST00000467281在不同样本中差异较大且表达值很低,不太符合实际情况。MRSeq方法通过L2/L1稀疏约束来考虑生物体所包含的生物特性。表2是MRSeq方法估计ENSG0000-00040597基因中6个异构体的表达水平。发现ENSG-000000040597基因表达主要由ENST00000000233、ENST00000463733和ENST00000489671异构体来体现,而ENST00000415666、ENST00000459680和ENST-00000467281异构体完全不表达。这符合基因发生表达时通常是由其包含的少数异构体所表现出来的生物特性。
为了进一步验证基因中相同异构体在不同技术性重复样本中其表达水平应该是相近的生物特性,本文选择小鼠数据集来验证异构体样本之间的表达水平。因为RNA-Seq测序技术的灵敏度很高,导致相关系数极容易受到少数高表达异构体的影响,为了避免这个问题,对表达水平对数化后再进行相关系数的计算。后续数据集的比较采用同样的相关系数计算方式。在表3中,MRSeq方法在大脑、肝脏和骨骼肌3个条件下都获得了比其他3个方法更好的结果,说明MRSeq方法能尽可能保持基因中相同异构体在不同样本中表达水平应该是相近的生物特性。
3.4qRT-PCR基因的表达水平验证
MAQC数据集因提供了约1 000个qRT-PCR验证基因,被最为广泛地用来比较各个方法的优劣性。通过映射到Ensembl注释库,最终匹配到833个基因,计算不同方法估计出的基因表达水平与qRTPCR验证的基因表达值之间的相关系数。由表4的结果可看出,MRSeq方法在单末端数据集上稍微优于Cufflinks和RSEM,但明显优于NURD,而在双末端数据集上,MRSeq方法的优势就较为明显。结果表明,MRSeq方法相比其他方法在基因表达水平估计上取得了较为准确的结果。
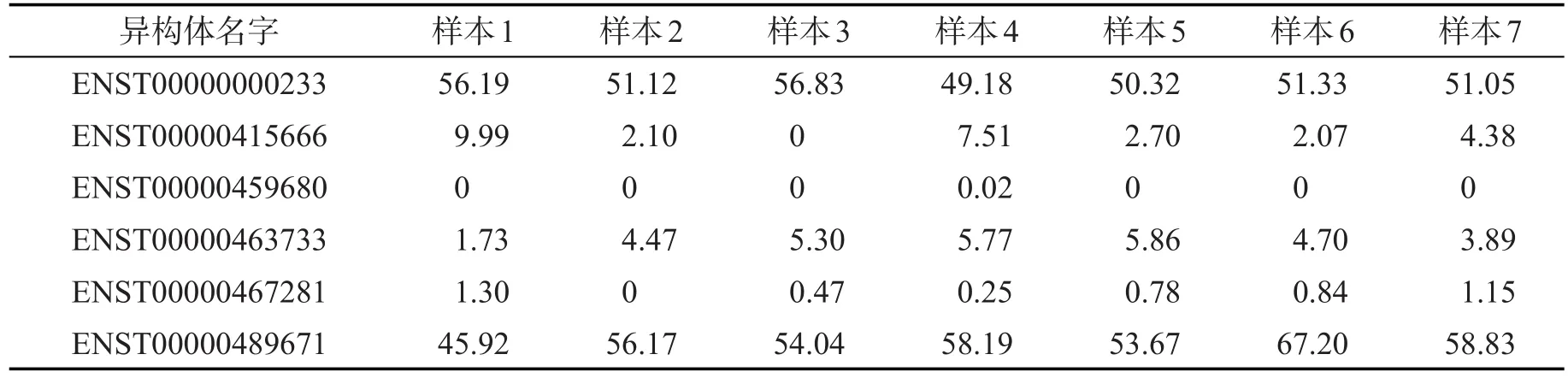
Table 1 Six isoforms expression of ENSG000000040597 gene estimated by Cufflinks in different samples表1 Cufflinks方法估计基因ENSG000000040597中6个异构体在不同样本中的表达水平
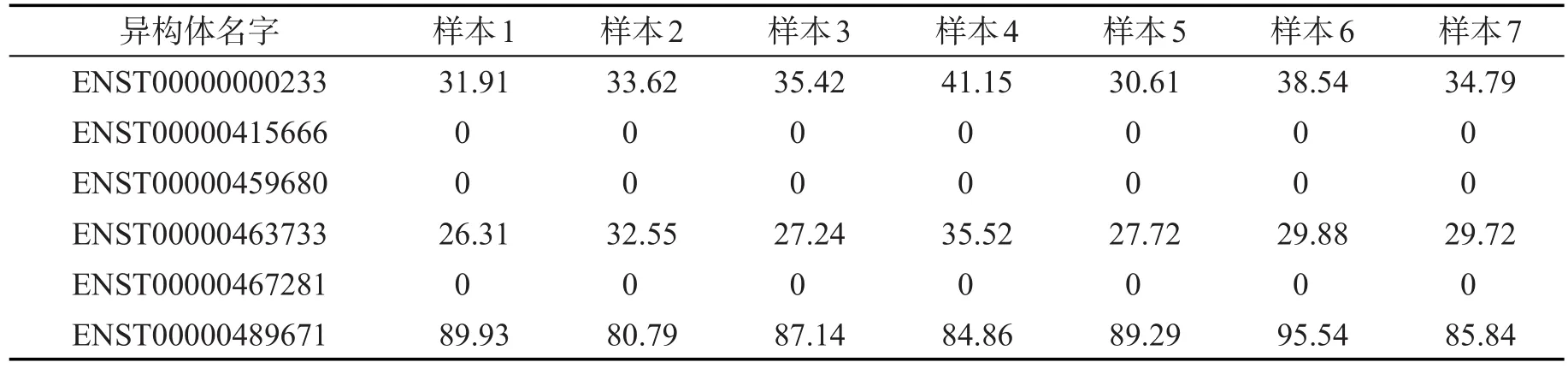
Table 2 Six isoforms expression of ENSG000000040597 gene estimated by MRSeq in different samples表2 MRSeq方法估计基因ENSG000000040597中6个异构体在不同样本中的表达水平
3.5qRT-PCR异构体表达水平验证
人类乳腺癌数据集被用来验证不同方法在异构体表达水平上的准确程度。文献[28]对8个异构体进行了qRT-PCR实验,其qRT-PCR值被作为标准值。计算不同方法估计出的异构体表达水平与qRT-PCR验证的标准值之间的相关系数。从表5中可看出,相比其他方法,MRSeq方法在两个不同条件下获得更高的相关系数。虽然在MCF-7条件上,4种方法的相关系数都非常低,但MRSeq方法仍能获得比其他方法更好的结果。结果表明,MRSeq方法在计算异构体表达水平上取得了较为准确的结果。
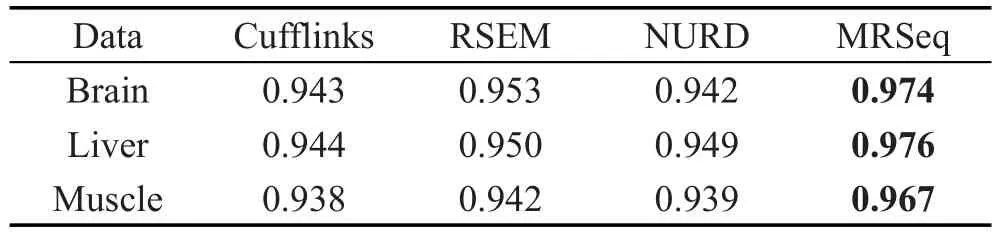
Table 3 Correlation coefficients of estimated isoform expression within samples by various methods in mouse dataset表3 小鼠数据集上各种方法估计的异构体表达水平在样本间的相关系数
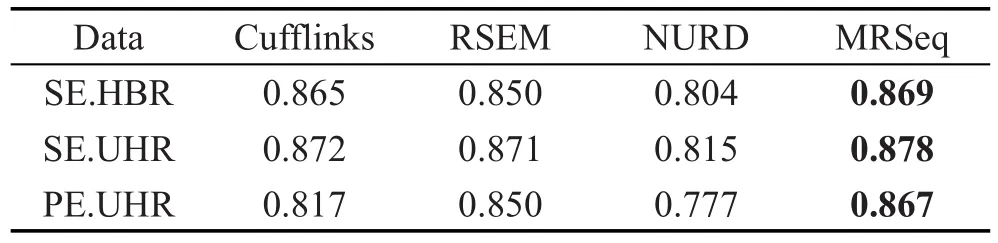
Table 4 Correlation coefficient of estimated gene expression by various methods with qRT-PCR results in MAQC dataset表4 MAQC数据集上各种方法与qRT-PCR验证基因间的相关系数
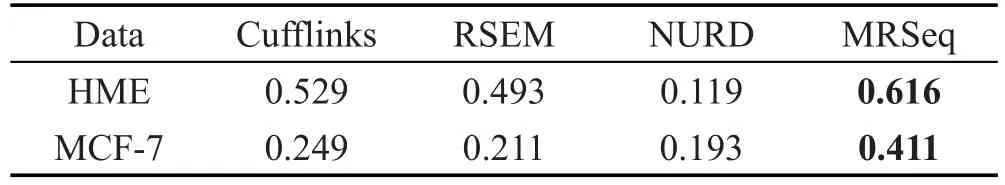
Table 5 Correlation coefficient of estimated isoform expression by various methods with qRT-PCR results in human breast dataset表5 人类乳腺癌数据集上各种方法与qRT-PCR验证异构体间的相关系数
3.6λ参数选择的分析
MRSeq方法通过L2 L1稀疏约束来考虑生物中的生物特性,参数λ的选择对表达水平计算有着很大影响,特别是异构体表达水平的计算。当λ→+∞,异构体在不同样本中都没有表达。随着λ的减小,基因中将会有异构体表达出来。选择MAQC数据集在HBR条件下的单末端数据集来分析不同λ的选择对结果产生的影响。图6(a)显示与qRT-PCR验证基因的相关系数,随着λ增大,相关系数有着明显下降,但是在区间[0,1]内,相关系数变化不大,表明λ的选择不应过大,较大的λ约束会导致一些真正的低表达基因或异构体被消除。而从图6(b)中可以看出,基因和异构体在样本之间的相关系数随着λ的增大有显著的升高,表明λ的增大能消除样本之间的技术性偏差而引起的噪声。为了权衡λ对表达水平计算的影响,根据在多个数据集上的结果,λ=1是个较优的选择,本文所有实验结果都采用此设置。
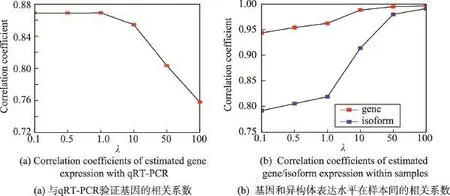
Fig.6 Effect ofλselection for expression estimation图6 不同λ选择对表达水平估计的影响
4 结论与展望
本文提出了一个基于多样本RNA-Seq数据的回归模型来估计基因和异构体表达水平,增加L2 L1稀疏约束来获得更好的生物解释。为了准确描述基因的读段分布在不同样本之间的高度相似性,设计了一个多样本数据的偏差曲线估计模型,通过多项回归模型获得偏差曲线,用来表示基因的读段分布在样本之间的共享特征。通过偏差曲线可以看出,基因的读段分布具有非均匀分布性质,在分布两端受到技术性和生物性偏差的影响,有明显的上升或下降的趋势,通过计算每个基因的偏差权重把读段分布的非均匀信息融入到表达水平的估计中。在小鼠数据集上,MRSeq方法估计的异构体表达水平在不同样本之间获得更高的相关系数,表示增加L2 L1稀疏约束可消除异构体在不同样本中受偏差和噪声而引起的差异,且保留了基因和异构体表达水平之间的稀疏性质,从而更好地保留了生物体中所包含的生物特性,获得了更好的生物解释性。在人类大脑和乳腺数据集上,通过与qRT-PCR基因和异构体的验证,MRSeq方法相比其他3种方法能获得更为准确的基因和异构体表达水平。在未来研究中,MRSeq方法基于已知的注释信息,但是生物体的注释信息远远没有达到完备状态,因此通过RNA-Seq测序数据来发现基因中未知异构体是很有意义的工作。而MRSeq方法可简单地扩展到此任务中,利用读段的跨结合区信息来预测可能存在的异构体,增加模型中基因和异构体的映射关系,通过L2 L1稀疏约束来寻找最有可能的未知异构体结构,将在后续的工作来验证此想法。
References:
[1]Marioni J C,Mason C E,Mane S M,et al.RNA-Seq:an assessment of technical reproducibility and comparison with gene expression arrays[J].Genome Research,2008,18(9):1509-1517.
[2]Margueat S,Bähler J.RNA-Seq:from technology to biology[J].Cellular and Molecular Life Sciences,2010,67(4): 569-579.
[3]Marguerat S,Wilhelm B T,Bähler J.Next-generation sequencing:applications beyond genomes[J].Biochemical Society Transactions,2008,36(5):1091-1096.
[4]Wang Zhong,Gerstein M,Snyder M.RNA-Seq:a revolutionary tool for transcriptomics[J].Nature Reviews Genetics, 2009,10(1):57-63.
[5]Burgess D J.Gene expression:a global assessment of RNA-seq performance[J].Nature Reviews Genetics,2014,15 (10):645-645.
[6]Jänes J,Hu Fengyuan,Lewin A,et al.A comparative study of RNA-seq analysis strategies[J].Briefings in Bioinformatics, 2015,16(6):1-9.
[7]Jiang Hui,Wong W H.Statistical inferences for isoform expression in RNA-Seq[J].Bioinformatics,2009,25(8):1026-1032.
[8]Hansen K D,Brenner S E,Dudoit S.Biases in illuminatranscriptome sequencing caused by random hexamer priming[J]. NucleicAcids Research,2010,28(12):e131.
[9]Risso D,Schwartz K,Sherlock G,et al.GC-content normalization for RNA-Seq data[J].BMC Bioinformatics,2011, 12(1):480.
[10]Huang Yan,Hu Yin,Jones C D,et al.A robust method for transcript quantification with RNA-Seq data[J].Journal of Computational Biology,2013,20(3):167-187.
[11]Ma Xinyun,Zhang Xuegong.NURD:an implementation of a new method to estimate isoform expression from non-uniform RNA-seq data[J].BMC Bioinformatics,2013,14(1):220.
[12]Wu Zhengpeng,Wang Xi,Zhang Xuegong.Using non-uniform read distribution models to improve isoform expression inference in RNA-Seq[J].Bioinformatics,2011,27(4):502-508.
[13]Hu Ming,Zhu Yu,Taylor J M G,et al.Using Poisson mixed-effects model to quantify transcript-level gene expression in RNA-Seq[J].Bioinformatics,2012,28(1):63-68.
[14]Srivastava S,Chen Liang.A two-parameter generalized Poisson model to improve the analysis of RNA-seq data[J]. NucleicAcids Research,2010,38(17):e170.
[15]Zhang Jing,Kuo C-C J,Chen Liang.WemIQ:an accurate and robust isoform quantification method for RNA-seq data[J]. Bioinformatics,2015,31(6):878-885.
[16]Trapnell C,Williams B A,Pertea G,et al.Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation[J].Nature Biotechnology,2010,28(5):511-515.
[17]Roberts A,Trapnell C,Donaghey J,et al.Improving RNASeq expression estimates by correcting for fragment bias[J]. Genome Biology,2011,12(3):R22.
[18]Li Bo,Ruotti V,Stewart R M,et al.RNA-Seq gene expression estimation with read mapping uncertainty[J].Bioinformatics,2010,26(4):493-500.
[19]Li Bo,Newey C.RSEM:accurate transcript quantification from RNA-Seq data with or without a reference genome[J]. BMC Bioinformatics,2011,12(1):323.
[20]Glaus P,Honkela A,Rattray M.Identifying differentially expressed transcripts from RNA-seq data with biological variation[J].Bioinformatics,2012,28(13):1721-1728.
[21]Zhang Li,Liu Xuejun,Chen Songcan.Detecting differential expression from RNA-seq data with expression measurement uncertainty[J].Frontiers of Computer Science,2015,9(4): 652-663.
[22]Behr J,Kahles A,Zhong Yi,et al.MITIE:simultaneous RNA-Seq-based transcript identification and quantification in multiple samples[J].Bioinformatics,2013,29(20):2529-2538.
[23]Zheng Xia,Wen Jianguo,Cheng Chung-Che,et al.NSMAP:a method for spliced isoforms identification and quantification from RNA-Seq[J].BMC Bioinformatics,2011,12(1):162.
[24]Jenatton R,Mairal J,Obozinski G,et al.Proximal methods for sparse hierarchical dictionary learning[C]//Proceedings of the 27th International Conference on Machine Learning, Haifa,Israel,Jun 21-24,2010.
[25]Langmead B,Salzberg S L.Fast gapped-read alignment with Bowtie 2[J].Nature Methods,2012,9(4):357-359.
[26]Mortazavi A,Williams B A,McCue K,et al.Mapping and quantifying mammalian transcriptomes by RNA-Seq[J].Nature Methods,2008,5(7):621-628.
[27]Canales R D,Luo Yuling,Willey J C,et al.Evaluation of DNA microarray results with quantitative gene expression platforms[J].Nature Biotechnology,2006,24(9):1115-1122.
[28]Wang E T,Sandberg R,Luo Shujun,et al.Alternative isoform regulation in human tissue transcriptomes[J].Nature, 2008,456(7221):470-476.

张礼(1985—),男,2010年于南京航空航天大学计算机应用专业获得硕士学位,现为南京航空航天大学博士研究生,主要研究领域为生物信息学,机器学习等。

刘学军(1976—),女,2006年于英国曼切斯特大学获得博士学位,现为南京航空航天大学计算机科学与技术学院教授,主要研究领域为生物信息学,机器学习等。

陈松灿(1961—),男,1997年于南京航空航天大学获得博士学位,现为南京航空航天大学教授、博士生导师,主要研究领域为机器学习,模式识别等。发表学术论文100余篇,主持国家自然科学基金、江苏省自然科学基金等多个项目。
Novel Method to Estimate Expression Level Based on Multi-Sample RNA-Seq Data*
ZHANG Li+,LIU Xuejun,CHEN Songcan
College of Computer Science&Technology,Nanjing University ofAeronautics&Astronautics,Nanjing 210016,China
+Corresponding author:E-mail:leo.zhang@nuaa.eud.cn
With the rapid development of the next-generation high-throughput sequencing technology,RNA-Seq has become the standard and important technique for transcriptome analysis.For multi-sample RNA-Seq data,the existing expression estimation methods usually deal with each single RNA-Seq sample,and ignore the read distributions with high consistency between multiple samples.This paper proposes a novel method,MRSeq,to estimate expression using multi-sample RNA-Seq data.MRSeq introduces a bias curve estimation model to capture the common features of read distributions shared among multiple samples.The common features are embedded into the model by deviation weight to correct read distributions.Meanwhile,by adding a sparse constraint,the method considers the sparsity between gene and the corresponding isoform expression.Three real datasets are used to validate the proposed method on gene and isoform expression estimation.Compared with the popular methods,MRSeq obtains more accurate gene and isoform expression estimation,and more meaningful biological explanation.
RNA-Seq;multi-sample;bias curve;sparse-specific;gene and isoform expression
2015-04,Accepted 2015-06.
ZHANG Li was born in 1985.He the M.S.degree in computer applications from Nanjing University of Aeronautics andAstronautics in 2010.Now he is a Ph.D.candidate at Nanjing University ofAeronautics andAstronautics.His research interests include bioinformatics and machine learning,etc.
LIU Xuejun was born in 1976.She the Ph.D.degree in computer science from University of Manchester, UK in 2006.Now she is a professor at College of Computer Science and Technology,Nanjing University of Aeronautics andAstronautics.Her research interests include bioinformatics and machine learning,etc.
CHEN Songcan was born in 1961.He the Ph.D.degree in communication and information systems from Nanjing University of Aeronautics and Astronautics in 1997.Now he is a Professor and Ph.D.supervisor at College of Computer Science and Technology,Nanjing University of Aeronautics and Astronautics.His research interests include machine learning and pattern recognition,etc.
10.3778/j.issn.1673-9418.1505045
*The National Natural Science Foundation of China under Grant No.61170152(国家自然科学基金);the Qinglan Project of Jiangsu Province(江苏省青蓝工程);the Fundamental Research Funds for the Central Universities of China under Grant No.CXZZ11_0217 (中央高校基本科研业务费专项资金).
CNKI网络优先出版:2015-06-18,http://www.cnki.net/kcms/detail/11.5602.TP.20150618.1646.001.html
A
TP391
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
浙江化工(2022年4期)2022-05-07
传染病信息(2021年6期)2021-02-12
学生天地(2020年6期)2020-08-25
数学年刊A辑(中文版)(2020年2期)2020-07-25
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
系统医学(2016年8期)2016-02-20
