
Alloying Effect Study on Thermodynamic Stability of MgH2by Firstprinciples Calculation
2016-11-24 07:31:13ZhenzhenWnZhongminWngDinhuiWngYnZhongJinqiuDengHuiyingZhouChohoHuSchoolofMterilsSciencendEngineeringGuilinUniversityofElectronicTechnologyGuilin541004ChinbGungxiExperimentCenterofInformtionScienceGuilin541004Chin
Zhen-zhen Wn,Zhong-min Wng,b∗,Din-hui Wng,Yn Zhong,b,Jin-qiu Deng, Hui-ying Zhou,Cho-ho Hu∗.School of Mterils Science nd Engineering,Guilin University of Electronic Technology,Guilin 541004,Chinb.Gungxi Experiment Center of Informtion Science,Guilin 541004,Chin
Alloying Effect Study on Thermodynamic Stability of MgH2by Firstprinciples Calculation
Zhen-zhen Wana,Zhong-min Wanga,b∗,Dian-hui Wanga,Yan Zhonga,b,Jian-qiu Denga, Huai-ying Zhoua,Chao-hao Hua∗
a.School of Materials Science and Engineering,Guilin University of Electronic Technology,Guilin 541004,China
b.Guangxi Experiment Center of Information Science,Guilin 541004,China
First-principles calculations based on density functional theory were performed to study the effect of alloying on the thermodynamic stability of MgH2hydride(rutile and fluorite structures)with transitional metals(TM=Sc,Ti,Y)and group IIA elements(M=Ca,Sr, Ba).The results indicate that fluorite structure of these hydrides are more stable than its relative rutile structure at low alloying content(less 20%),structural destabilization of MgH2appears in the alloying cases of Ti,Sr and Ba respectively.The structure-transition point from rutile structure to fluorite structure is at around 20%for MgH2-TM,and about 40%for MgH2-M.The formation enthalpy of fluorite Mg0.5Ba0.5H2is about 0.3 eV and higher than that of fluorite MgH2,indicating that its hydrogen-desorption temperature at atmospheric pressure will be much lower than that of pure MgH2.Good consistency between experimental and calculated data suggests that above-adopted method is useful to predict structural transition and properties of MgH2based hydrides for hydrogen storage.
MgH2,First-principles study,Alloying,Destabilization,Structural transition
I.INTRODUCTION
As one of the most promising hydrogen storage materials,magnesium hydride(MgH2)has attracted huge interest in hydrogen storage field due to its abundant resource,low cost and high hydrogen-storage capacity of 7.6 wt%.However,its application in hydrogen-storage is limited because of its poor hydrogen absorption and desorption performance.In practice it often takes several hours for(de)hydrogenation at a relatively higher temperature about 623 K[1−3].
To improve the hydrogenation kinetics of Mg hydride, a large number of experimental and theoretical investigations have been performed extensively in the last decades.These studies have shown that doping the third foreign elements into MgH2is an efficient way to decrease dehydrogenation temperature and expedite kinetics of MgH2[4−6].Kelkar and co-authors have found that the Mg−H bond in Al-doped MgH2is more susceptible to dissociation and thus the thermodynamic stability of MgH2is decreased[7,8].The TMs(Ti,Mn, and Ni)doping influence on(110)surface of MgH2has been investigated by Dai et al.[9,10].They have found that Ti atoms prefer to occupy both substitutional[9]and interstitial sites[10],while Mn and Ni tend to occupy the interstitial sites near the surface of MgH2.An investigation performed by Er et al.[11]has shown that for TM(TM=Sc,Ti,V,and Cr)concentration approaching x=0.2 in MgxTM1−xH2,the fluorite structure with cubic H environment becomes more stable than the rutile one.Mamula et al.have investigated electronic structure and charge distribution topology of MgH2doped with 3d transition metals using the full potential(linearized)augmented plane waves method with addition of local orbitals(FP-LAPW+LO and APW+LO),and found that along the 3d series TMs accomplish different kinds of bonding with the nearest and next-nearest neighbor hydrogen atoms that in general weaken related Mg−H bonds and destabilize the surrounding MgH2matrix[12].
A major improvement in discharge kinetics resulting from alloying Mg with Sc has been reported by Notten et al[13].Up to 80%of Mg contents,the reversible capacity exceeded 5 wt%and the corresponding desorption properties were still excellent.The Mg-Sc system has been extensively studied by using several diffraction techniques,and it was found that the ternary hydride retains the fluorite-type structure of pure ScH2[14].This structure contains large empty octahedral interstitials,which facilitate rapid hydrogen diffusion.This has recently been confirmed by Conradi et al.[15]. NMR measurements have shown that the hopping rates of hydrogen between different hydrogen sites in ScH2and Mg0.65Sc0.35H2are about seven and eight orders of magnitude more rapid than that in MgH2,respectively. Recently,based on the theoretical considerations presented by Vajeeston et al.[16]and their experimental finding[17],Pauw and co-authors[18]have further concluded that for MgH2the transformation from a rutiletype structure to a fluorite-type structure could be accelerated by alloying with transition metals.
∗Authors to whom correspondence should be addressed.E-mail: zmwang@guet.edu.cn,chaohao.hu@guet.edu.cn
In this work,based on two crystallographic modifications of MgH2(rutile-type and fluorite-type),a theoretical investigation based on density functional theory(DFT)of MgH2-transitional metals(TM=Sc,Ti, Y)and MgH2-group IIA elements(M=Ca,Sr,Ba)was carried out.The destabilizing mechanism of MgH2alloying with TM/M for hydrogen storage performance was also discussed via analyzing the change in the calculated enthalpy of formation,lattice parameters,and electron density of states(DOS).
II.COMPUTATIONAL DETAILS
All DFT calculations were performed using the Vienna ab initio Simulation Package[19]. The interactions between core and valence electrons were described with the projector augmented wave method[20]. Perdew-Burke-Ernzerhof implemented generalized gradient approximation[21]was used to treat the exchange and correlation energies.The plane-wave energy cutoff was set to 400 eV and the k-point mesh in the Brillouin zone was about 0.03×2π˚A−1in all calculations. During structure optimization the lattice parameters, volume and atom positions were allowed to relax fully within symmetry restrictions.The convergence criterion for total energy was set to 10−5eV during the self-consistent calculations.
Two types of crystal structure of MgH2which are the rutile structure(tetragonal,P42/mnm)and fluorite structure(cubic,Fm-3m)are generally considered. In this work,the super cell models containing 8 metal atoms(seen in Fig.1)were established in order to consider the effect from different alloying contents on the formation enthalpy of the two structural modifications.
In general,the stability of any compound can be evaluated by its formation enthalpy.The formation enthalpy of MgH2based hydrides can be defined as:
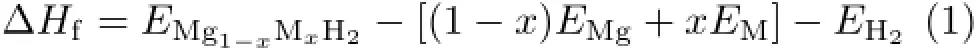
where EMg1-xMxH2,EMgand EMare the calculated total energies of Mg1−xMxH2,Mg,and M bulk materials, and EH2is the energy of isolated H2molecule.
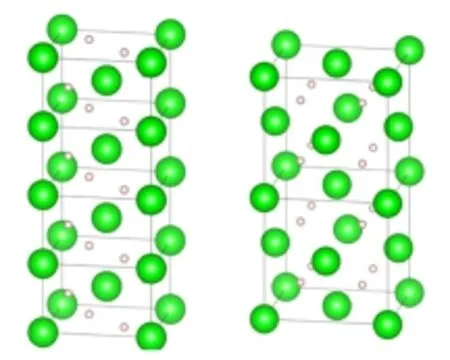
FIG.1 1×1×4 cell of the rutile structure(left,tetragonal, P42/mnm)and 1×1×2 cell of the fluorite structure(right, cubic,Fm-3m).The big spheres are Mg atoms,and the small spheres are H atoms.
III.RESULTS AND DISCUSSION
A.MgH2-TM system hydrides
Calculated formation enthalpies of MgH2-TM (TM=Sc,Ti,Y)hydrides are shown in Fig.2.The calculated formation enthalpy of rutile MgH2is−0.64 eV (or−61.8 kJ/mol),which has about 20%difference compared with the literature value(−77 kJ/mol)[22], but agrees well with the theoretical results(−0.66 eV) [13,16−18]. Considering that the experiment was carried out under 673 K,so these calculated values at ground state are acceptable.
In the case of MgH2with rutile structure,the formation enthalpies decrease with the increase of alloying content of Sc and Y,indicating that rutile structure of MgH2is stabilized by alloying with Sc or Y. While an apparent increase of formation enthalpy is obtained when alloying with Ti in the range of 0−37.5%, Mg0.625Ti0.375H2has a higher formation enthalpy value of−0.45 eV,which is 0.19 eV higher than that of pure MgH2,but the formation enthalpy will decrease when the alloying content of Ti is above 37.5%.
In the case of MgH2with fluorite structure,the structure of pure MgH2is less stable than the rutile structure of MgH2.But the fluorite structure becomes more stable than the rutile structure when Sc,Ti or Y atoms doping content reaches 20%,which indicates that Mg0.8TM0.2H2energetically prefers to a cubic fluorite structure.This is consistent with the experimental findings suggested by X-ray diffraction[13,14].It also agrees well with the results of electrochemical experiments,which has reported that alloying with 20% of transitional metal can greatly improve the hydrogen desorption kinetic performance[23].
Figure 2 shows an important point,20%of alloying content is a structure transition point from rutile structure to fluorite structure,the formation enthalpy of fluorite structure of MgH2becomes more negative than that with rutile structure when x is above 0.2. As to Mg1−xTixH2calculation,the rutile structure is destabilized by 0.17 eV when x=0.25.The most unsta-ble status is around x=0.375,the formation enthalpy of Mg0.5Ti0.5H2is−0.39 eV,which is 0.24 eV more positive than that of pure MgH2.However,the fluorite structure still becomes the most stable one at x=0.2,similar to Mg-Sc system.These results are in accordance with the results reported by Pauw et al. [18].MgH2-TM system hydrides have the structuretransition tendency from rutile structure to fluorite structure at a specific point of alloying content,the transition point is about 20%for transition metals.

FIG.2 Formation enthalpy of Mg-TM hydrides.
B.MgH2-M system hydrides
Calculated formation enthalpiesofMg1−xMxH2(M=Mg,Ca,Sr)are shown in Fig.3.It can be clearly found from Fig.3 that alloying with Ca will result in the increase of thermodynamic stabilities of Mg1−xCaxH2hydride since the calculated formation enthalpies of the rutile and fluorite structure always decrease with the increase of alloying content.However,the alloying treatment with Sr can induce a slight decrease in the thermodynamic stabilities of rutile Mg1−xSrxH2,since the enthalpy of Mg0.625Sr0.375H2is 0.06 eV higher than that of MgH2.Figure 3 also clearly indicates that the most obvious decrease in structural stability of both rutile and fluorite structure is from the alloying treatment with Ba.It can be observed that the enthalpy of rutile Mg0.5Ba0.5H2is 0.33 eV higher than that of MgH2. In addition,the structural transition point from the rutile to fluorite structure is around 40%for Mg1−xMxH2,which has a big delay compared with that of Mg1−xTMxH2hydrides(about 20%).At this point, its theoretical hydrogen-storage capacity is 3.1 wt%.
C.Electronic properties of Mg0.5Ba0.5H2
As mentioned above,alloying with Ba will lead to an obvious decrease in the thermodynamic stability of Mg1−xMxH2and the fluorite Mg0.5Ba0.5H2has a high formation enthalpy,so here Mg0.5Ba0.5H2has been selected to make a comparison with pure MgH2.After structure optimization,the cubic fluorite structure of Ba doped MgH2is distorted,and the unit cell volume is obviously increased by 39.9%.The interatomic distance between Mg and H atoms also significantly increased from 1.94˚A to 2.24˚A.So the interaction between Mg and H will be weakened.Calculated DOS of rutile MgH2is shown in Fig.4.There is a wide band gap(about 2.73 eV)between the conduction band and valence band,revealing its insulating nature.The partial DOS of MgH2suggests that its valence bands are mainly contributed by H 1s states,while the contribution from the Mg 3s and 2p orbitals is not so much.The involvement of 2p electrons in the valence band is an evidence of hybridization of Mg 3s and 2p orbitals.The DOS of Ba-doped MgH2(Mg0.5Ba0.5H2)is presented in Fig.5.With Ba-doping,the Ba 5p and 4d orbitals make a distinct contribution to the valence band.While a peak within the energy range from−10 eV to−15 eV is mainly ascribed to the hybridization of H 1s and Ba 5p orbitals,indicating that there exists a strong bonding between Ba and H atoms.Although Mg0.5Ba0.5H2still keeps its non-metallic nature,its metallization is obviously improved as suggested from the relative decrease of band gap.

FIG.3 Formation enthalpy of Mg-M hydrides.
IV.CONCLUSION
Based on two crystallographic modifications of MgH2(rutile-type and fluorite-type),the relative thermodynamic stabilities of MgH2-TM and MgH2-M(TM=Sc, Ti,Y,M=Ca,Sr,Ba)have been studied by firstprinciples calculations.Rutile structure of MgH2will transform into fluorite structure when alloying with these elements.For MgH2-TM hydrides,the structuretransition point from rutile structure to fluorite structure is at around 20%substitution,which agrees very well with both experimental and previous calculated data. For MgH2-M hydrides,this structural transition point is at around 40%substitution. Further-more,Ti Sr,and Ba substitution not only cause the structure transition,but also reduce its thermodynamic stabilities,especially in the case of Ba substitution. The formation enthalpy of fluorite Mg0.5Ba0.5H2is 0.33 eV higher than that of pure rutile MgH2,suggesting that its hydrogen-desorption temperature at atmospheric pressure will be much lower compared to pure MgH2.Mg0.5Ba0.5H2also has a reasonable theoretical hydrogen-storage capacity of about 3.1 wt%,which indicates that it would be a potential candidate of hydrogen storage materials.
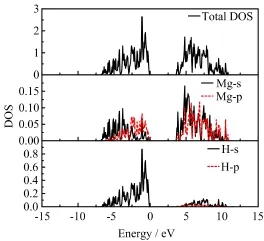
FIG.4 Calculated DOS of rutile MgH2.
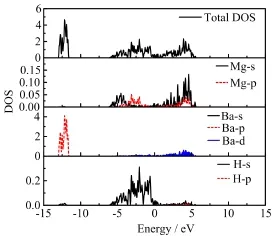
FIG.5 Calculated DOS of fluorite Mg0.5Ba0.5H2.
V.ACKNOWLEDGMENTS
This work is supported by the National Natural Foundations of China(No.51261003,No.51471055, No.51401060,and No.11464008),the Natural Foundations of Guangxi Province(No.2016GXNSFGA380001 and No.2014GXNSFGA118001),and Guangxi Experiment Center of Information Science(No.20130113 and No.YB1512).
[1]J.Huot,G.Liang,S.Boily,A.Van Neste,and R. Schulz,J.Alloys Compd.293,495(1999).
[2]A.Ranjbar,Z.P.Guo,X.B.Yu,D.Wexler,A.Calka, C.J.Kim,and H.K.Liu,Mater.Chem.Phys.114, 168(2009).
[3]X.Luo,D.M.Grant,and G.S.Walker,J.Alloys Compd.622,842(2015).
[4]M.Abdellatief,R.Campostrini,M.Leoni,and P. Scardi,Int.J.Hydrogen Energy 38,4664(2013).
[5]N.E.Galushkin,N.N.Yazvinskaya,and D.N. Galushkin,ECS Electrochem.Lett.2,A1-2(2013).
[6]S.C.Zhou,R.K.Pan,T.P.Luo,D.H.Wu,L.T. Wei,and B.Y.Tang,Int.J.Hydrogen Energy 39,9254 (2014).
[7]T.Kelkar and S.Pal,J.Mater.Chem.19,4348(2009).
[8]T.Kelkar,S.Pal,and D.G.Kanhere,Chem.Phys. Chem.9,928(2008).
[9]J.H.Dai,Y.Song,and R.Yang,Int.J.Hydrogen Energy 36,12939(2011).
[10]J.H.Dai,Y.Song,and R.Yang,J.Phys.Chem.C 114,11328(2010).
[11]S.Er,D.Tiwari,G.A.Wijs,and G.Brocks,Phys.Rev. B 79,024105(2009).
[12]B.P.Mamula,J.G.Novakovi´c,I.Radisavljevi´c,N. Ivanovi´c,and N.Novakovi´c,Int.J.Hydrogen Energy 39,5874(2014).
[13]P.H.L.Notten,M.Ouwerkerk,H.van Hal,D.Beelen, W.Keur,and J.Zhou,J.Power Sources 129,45(2004).
[14]W.P.Kalisvaart,R.A.H.Niessen,and P.H.L.Notten, J.Alloy Compd.417,280(2006).
[15]M.S.Conradi,M.P.Mendenhall,T.M.Ivancic,E.A. Carl,C.D.Browning,and P.H.L.Notten,J.Alloy Compd.447,499(2007).
[16]P.Vajeeston,P.Ravindran,A.Kjekshus,and H. Fjellv˚ag,Phys.Rev.Lett.89,175506(2002).
[17]P.Vajeeston,P.Ravindran,B.C.Hauback,H.Fjellv˚ag, A.Kjekshus,Furuseth,and M.Hanfland,Phys.Rev.B 73,224102(2006).
[18]B.R.Pauw,W.P.Kalisvaart,S.X.Tao,M.T.M. Koper,A.P.J.Jansen,and P.H.L.Notten,Acta Materialia 56,2948(2008).
[19]G.Kresse and J.Furthm¨uller,Phys.Rev.B 54,11169 (1996).
[20]P.E.Blochl,Phys.Rev.B 50,17953(1994).
[21]J.P.Perdew,K.Burke,and M.Ernzerhof,Phys.Rev. Lett.77,3865(1996).
[22]R.Griessen and T.Riesterer,Heat of Formation Models,In: L.Schlapbach Ed.,Berlin: Springer, (1988).
[23]K.Miwa and A.Fukumoto,Phys.Rev.B 65,155114 (2002).
(Dated:Received on February 29,2016;Accepted on May 12,2016)
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年5期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年5期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Preparation of Bio-hydrogen and Bio-fuels from Lignocellulosic Biomass Pyrolysis-Oil
- Combination Computing of Support Vector Machine,Support Vector Regression and Molecular Docking for Potential Cytochrome P450 1A2 Inhibitors
- Working Condition Real-Time Monitoring Model of Lithium Ion Batteries Based on Distributed Parameter System and Single Particle Model
- Hydrodeoxygenation of Anisole over Ni/α-Al2O3Catalyst
- Highly Efficient and Selective Removal of Pb(II)ions by Sulfur-Containing Calcium Phosphate Nanoparticles
- Efficient Removal Phenol Red over Ternary Heterostructured Ag-Bi2MoO6/BiPO4Composite Photocatalyst