
Photodissociation Study of Ca+-Dipropylamine Complex
2016-11-24 07:31:13XiyaChenBingJinYuanChengDongshengWangShiheYangaSchoolofElectricalandElectronicEngineeringHuazhongUniversityofScienceandTechnologyWuhan430074ChinabStateKeyLaboratoryofMagneticResonanceandAtomicandMolecularPhysicsWuhan
Xi-ya Chen,Bing Jin,Yuan Cheng,Dong-sheng Wang,Shi-he Yanga.School of Electrical and Electronic Engineering,Huazhong University of Science and Technology, Wuhan 430074,Chinab.State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,Chinac.State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,Chinad.Shenyang Branch,Chinese Academy of Sciences,Shenyang 110004,Chinae.Department of Chemistry,The Hong Kong University of Science and Technology,Clear Water Bay, Kowloon,Hong Kong,China
Photodissociation Study of Ca+-Dipropylamine Complex
Xi-ya Chena,b,Bing Jinb,c,Yuan Chengb,c,Dong-sheng Wangd∗,Shi-he Yange∗
a.School of Electrical and Electronic Engineering,Huazhong University of Science and Technology, Wuhan 430074,China
b.State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,China
c.State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China
d.Shenyang Branch,Chinese Academy of Sciences,Shenyang 110004,China
e.Department of Chemistry,The Hong Kong University of Science and Technology,Clear Water Bay, Kowloon,Hong Kong,China
Gas-phase Ca+-dipropylamine complex has been subjected to photodissociation (400−690 nm).One reactive product,Ca+-NHC3H7,is detected only in the region of 450−528 nm,besides the evaporation fragment of Ca+,which is dominant throughout the whole spectral region we have studied.The photoreaction channel may be explained in terms of a metal insertion mechanism.The calculated results are in good agreement with the experimental observations.
Photodissociation,Ca+,Dipropylamine,B2-PLYP,TD-DFT
I.INTRODUCTION
Metal ion-ligand systems play an important role in a wide range of chemical,biological and atmospheric processes[1−11]. Photodissociation spectroscopy of metal ion-ligand complexes is a useful means of exploring their photo-induced reactions.Singly charged alkaline earth metal ions are ideal for such photodissociation studies by virtue of their substantial oscillator strengths in the visible and ultraviolet spectral regions,which can be easily accessed by pulsed laser sources[3,4,6,12−14].During the last few years, many photo-induced reactions of Mg+-ligand complexes have been studied experimentally,for example,those of Mg+-XCH3(X=H,F,Cl,I)[1,15,16],Mg+-H2O [17−19],Mg+-CH3OH[20,21],Mg+-acetone,Mg+-XHCO(X=H,CH3)[22,23],Mg+-nitromethane[24], Mg+-dioxanes[25],Mg+-NH3[26],Mg+-diethylamine [27],Mg+-triethylamine[27],Mg+-NCCH3[28],Mg+-NCSR(R=CH3,C2H5)[29],Mg+-XCNC2H5(X=O,S) [29,30],Mg+-2,2,2-trifluoroethanol[31],Mg+-C6H5X (X=H,F,Cl,Br)[32],Mg+-(o-,m-,p-C6H4F2)[33], Mg+(2-fluoropyrdine)[34],Mg+-thymine and Mg+-uracil[35].In the ground states of such complexes,Mg+is always coordinated to highly electronegative O,F,or N atoms,and the electrostatic interaction is dominant. Upon excitation,the Mg+ion would induce photoreactions by inserting into C−X(X=N,F,O)bonds.
A variety of complexes of Ca+-ligand complexes have been studied experimentally[36−42]. These studies have indicated that little of reactive product in the visible region,suggesting Ca+is less reactive in excited states than Mg+.For example,in M+-CH4clusters, excited Mg+efficiently yields the product MgCH3+through a C−H bond insertion reaction[13,43],while photo-excited Ca+-CH4merely dissociates through an evaporation channel[40].Recently,we have studied the photodissociations of Ca+-tert-buylamine[44]and Ca+-pyridine[45].In both cases,we found a photoinduced channel leading to the reactive product Ca+-NH2.In this work,we report on the photoreaction of the Ca+-dipropylamine complex.We detected the reactive product Ca+-NHC3H7in the region 450−528 nm. Quantum ab initio calculations have been carried out to access the ground-state structure,binding energies,and reaction energies.The possible photoreaction mechanisms are discussed.Our theoretical calculations are in good agreement with all of the observed spectral features.
II.EXPERIMENTS AND CALCULATIONS
The experiment was performed in a cluster apparatus equipped with a reflectron time-of-flight mass spectrometer(RTOFMS).Details of the main experimental setup have been provided previously[33,46].Hence, only the aspects relevant to the present experiment are described here.A rotating CaO plate(5 mm in diameter)was mounted 15 mm downstream from the exit of a pulsed valve(General Valve,Series 9).The second harmonic(532 nm,~1 mJ/pulse)of a Nd:YAGlaser(Continuum,Surelite I)was weakly focused on a spot on the CaO plate to generate Ca+ions.The CaO plate was rotated by a step motor upon each laser pulse to expose a fresh surface during the laser-ablation experiments.Dipropylamine vapor was seeded in He at a backing pressure of 10 psi expanded through a 0.5 mm diameter orifice of the pulsed valve.The supersonic jet molecular beam traversed perpendicularly to the laser-produced species containing metal ions and atoms 20 mm from the CaO plate,and formed a series of metal cations solvated by dipropylamine.The nascent complexes traveled 140 mm downstream into the extraction region of the RTOFMS.The complexes were accelerated vertically by a high voltage pulse in a two-stage extractor.After extraction,the complexes were steered by a pair of horizontal plates and a pair of vertical deflection plates.Desired complex cations were selected by a two-plate mass gate,then entered into the reflectron after a long free potential transit.Upon arrival at the turn-around region of the reflectron,selected cations were irradiated with the collimated beam of a tunable dye laser(ScanMate II,Lambda Physik) pumped by a Nd:YAG laser(Continuum Powerlite Precision II,8000 series).The parent and nascent daughter cations were re-accelerated by the reflectron electric field and detected by a dual-plate microchannel plate detector(MCP).The photolysis laser fluence was kept low(<1 mJ/cm2)to avoid multiphoton processes and saturation phenomena.
∗Authors to whom correspondence should be addressed.E-mail: chsyang@ust.hk,dswang@mail.syb.ac.cn
Quantum mechanical calculations were performed using the Gaussian 09 program[47].The observed photoreactions,although initiated by electronic excitation, were assumed to occur on the ground-state surface after a series of internal conversions,the reliability of which has been confirmed for many systems[16,24, 25,30,32,33,35,46].All species involved were optimized using the double-hybrid functional B2-PLYP method.The nature of each stationary point was characterized by vibrational frequency calculations(minimum with zero imaginary frequency,transition state with one imaginary frequency).Intrinsic reaction coordinate(IRC)calculations were also performed to confirm the transition structures.The vertical excitation energies and corresponding oscillator strengths were calculated by the TD-DFT/B2-PLYP method on each optimized ground-state geometry.The SDD pseudopotential and basis sets for Ca and the 6-31++G(2df,pd) basis sets for C,H and O atoms were employed in all of the calculations[14].All relative energies are reported with zero-point energy corrections(ZPE).All calculations turned off the use of symmetry in the CPHF equations.

FIG.1 Photodissociation difference massspectra of Ca+-NH(C3H7)2at(a)460 nm and(b)530 nm.The negative peaks represent parent complexes while the positive ones relate to photofragments.
III.RESULTS AND DISCUSSION
A.Photoreaction channels
Typical photodissociation difference mass spectra of Ca+-NH(C3H7)2at 460 and 530 nm are shown in Fig.1, where the negative peaks represent the parent complexes while the positive relates to the photofragments. In the long wavelength region,only one photofragment, the Ca+ion,is produced. In the short wavelength region,a very small peak indicates the production of the reactive photofragment Ca+-NHC3H7,accompanying the elimination of the C3H7radical.The photofragments can be summarized in terms of the reactions as follows:
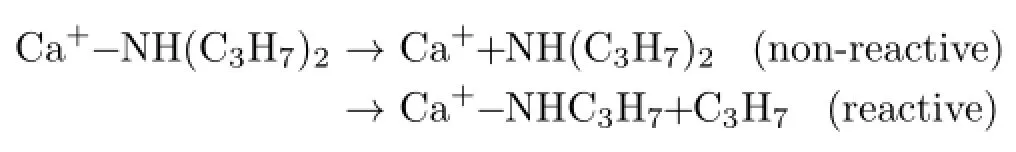
At both photon energies,the major photofragment, Ca+,is dominant,and can be considered to arise from evaporation photodissociation upon electronic excitation.The observation of the reactive product Ca+-NHC3H7only in short-wavelength indicates that the barrier of the photo-reaction and the appearance energy of the Ca+-NHC3H7is very high,which is due to the strong steric hindrance.Meanwhile,in competition with non-reaction pathway,the high barrier of the reactive pathway makes photoproducts have a small branching fraction.Therefore,the intensity of the reactive product in Fig.1 is very small.Furthermore,compared with the rich photo-induced reaction patterns observed for complexes containing Mg+,it is evidently relatively difficult to induce photoreaction in the excited Ca+-ligand complex.
B.Structural and energetic analysis of the parent complex and photoproducts
Figure 2 displays the optimized structures of the complex and its photoproducts,along with the calculated energies,bond lengths and atomic charges on Ca+.The most stable structure of the complex is characterized by the linkage of Ca+to the N atom,which suggeststhat the charge-dipole moment interaction is dominant [1−4].The energy of GS1 is considered to be the zeropoint of the energy in the following sections.In the ground state,GS2 is another stable structure of the complex and is 1.47 eV higher in energy than GS1. However,GS2 has a Ca+insertion structure and then it must be formed via the Ca+-insertion transition state, where higher energy is required.In our cluster sources, it must have high energy and directly dissociate into Ca+-NHC3H7product and C3H7radical if GS2 could be formed.Furthermore,the binding energy of the GS2 is about 0.93 eV which is smaller than that of the GS1. If there was a small amount of GS2 in parent ions,the reactive product Ca+-NHC3H7would be observed in longer wavelength.Therefore,the most stable GS1 is considered to the only parent ion.The formation of the reactive product Ca+-NHC3H7and the elimination of the C3H7radical is indicative of operation of the metal insertion mechanism[1,13,40,48].The dissociation energies for the non-reactive channel and reactive channel are estimated to be 1.67 and 2.40 eV,respectively. The respective channels are discussed in detail in the following section.

FIG.2 Optimized structures of the complexes Ca+-NH(C3H7)2and photoproducts.The bond lengths are given in˚A.The energies below represent the calculated appearance energies of the corresponding species.
C.Action spectra and photofragment branching curve
Figure 3 shows the photodissociation action spectrum of the total ion yield of the photofragments in the wavelength range of 400−690 nm.The spectrum consists of four bands,which are all situated on the red side of the2D←2S atomic transition of Ca+.The four peaks are centered at 610,570,502,and 432 nm,respectively.It can clearly be seen that all of the peaks are structureless,and that the three peaks at longer wavelength are significantly stronger than that at 432 nm. Clearly,these bands result from splittings of the electronic transitions of Ca+(2D←2S,2P←2S)in the presence of dipropylamine.

FIG.3 Photodissociation action spectrum ofCa+-NH(C3H7)2.Points indicate experimental results.Solid lines indicate theoretically calculated vertical transition energies for the complex.
The vertical transition energies of the complex are depicted in Table I.The eight lowest-lying excited states have been calculated.The first five transitions at 748.4, 737.0,679.0,665.6,and 599.9 nm are attributed to splittings of the Ca+-based2D←2S transition,and the three higher transitions at 500.1,434.6,and 285.4 nm are due to Ca+-based2P←2S.The order of these excited states results from the different electron populations of each orbital,which has been discussed extensively in other scientific literature.Because the Ca+-based 3dxy,3dx2−y2←4s transition shows only very weak absorption and the 4pz←4s transition lies far away in the ultraviolet region,we selected only the wavelength range 400−690 nm to study the photo-induced reaction.For ease of comparison with the experimental findings,the relevant calculation results are also presented in Fig.3.The calculations indicate that the bandcentered at 610 nm consists of two transitions of Ca+-based 3dxy←4s and 3dxz←4s,and that the band at around 570 nm is assigned to the transition of Ca+-based 3dz2←4s.These transitions are due to splittings of the Ca+(2D←2S)atomic transition and are blueshifted to it.Molecular orbital analysis indicates that these blue-shift features are attributed to the aforementioned transition mixed with the transitions to states 4py,4px,and 4pz,respectively.Higher level calculations would be desirable to get more accurate results. However,the current study did not make such effort since our calculations have given the reliable results of assigning the experimental bands.The observed bands centered at 502 and 432 nm are assigned to the Ca+-based 4py←4s and 4px←4s transitions,respectively. The action spectra of the complex are consistent with TD calculations on the peak positions(see Fig.3),which supports the proposed structure of the complex in which Ca+is linked to the N atom and also confirms our assignments of the observed transitions.

TABLE I Electronic transition positions(nm),oscillator strengths and assignments for the eight lowest-lying excited states of the Ca+-NH(C3H7)2complex.Theoretical results were obtained using TD-B2-PLYP with SDD pseudopotential and basis sets for Ca and 6-31++G(2df,pd)basis sets for C,H,and O atoms.
D.Photofragment branching fractions
Figure 4 presents the branching fractions of the observed fragments as a function of photolysis laser wavelength.The data in Fig.4 show that the non-reactive product Ca+is the only photoproduct over the whole range except for the region 450−528 nm.In this small region,corresponding to the transition of Ca+-based 4py←4s,the reactive photoproduct Ca+-NHC3H7is observed.The amount of Ca+-NHC3H7is very small and the branching fractions of the photoproducts are nearly constant at about 0.03,giving the impression that the photoreaction channel shows remarkable electronic state-specificity.Combined with the action spectroscopy,however,it can be concluded that a much lower intensity of the band around 432 nm makes the reactive channel impossible to observe.Therefore,we do not consider this reactive channel to show an electronic state-specificity.

FIG.4 Photofragment branching fractions from Ca+-NH(C3H7)2as a function of the laser wavelength.

FIG.5 Reaction coordinate for the photoprocesses of Ca+-NH(C3H7)2. Pathway I(blue lines)is an evaporationdissociation channel.In pathway II(red lines),the metal insertion is responsible for the reaction channel.
E.Formation of the photoproduct
The photo-induced evaporation and reaction pathways of the title complex are summarized in Fig.5,along with the calculated energies.Pathway I(blue lines)is an evaporation-dissociation channel.After a series of pre-dissociations,the excited complex GS1 dissociates directly into a free Ca+cation and dipropylamine,requiring an energy of just 1.67 eV.For pathway II(red lines),the metal insertion is responsible for the reaction channel[1,48].Upon transition to the 4pyorbital, the complex absorbs a photo energy of 2.48 eV,which is sufficient for the ensuing photoreactions.First,the excited parent ions returned to the ground state after a series of non-adiabatic interactions.The next step is the insertion of Ca+into the Cα−N bond,forming the insertion structure(GS2)via the transition state (TS1),which is 2.30 eV less stable than GS1.GS2 is a stable structure that is 1.47 eV higher in energy than GS1.Finally,GS2 dissociates into two photofragments, Ca+-NHC3H7and C3H7.The calculated appearanceenergy of the Ca+-NHC3H7is 2.40 eV(516 nm)which approximately matches the observed value of 2.35 eV (528 nm).This excellent agreement suggests that our calculations are reliable and that the insertion mechanism is reasonable for this band[1,48].For the transition to the higher 4pxorbital,the low intensity results in the reactive product not being seen experimentally.
IV.CONCLUSION
Complexes containing Ca+and NH(C3H7)2have been produced in a supersonic beam.By mass-selecting the Ca+-NH(C3H7)2complex,we have measured the relative photodissociation product yields as a function of the excitation wavelength in the region 400−690 nm. The reactive photoproduct Ca+-NHC3H7has been identified in low yields compared to the dominant evaporation photofragment Ca+.Quantum mechanical calculations have been performed to obtain the groundstate structures and energetics of the parent complexes and daughter photoproducts.In the ground-state structure of the complex,Ca+is linked to the N atom,and the characteristic charge-dipole interaction is dominant. The photoreaction channel is explained well by our calculations based on Ca+insertion mechanism[1,48]. We also suggest that the failure to observe the reactive product in the high energy band is due to its low intensity signal in that region.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.11504245).
[1]P.D.Kleiber and Chen,J.Int.Rev.Phys.Chem.17, 1(1998).
[2]C.S.Yeh,J.S.Pilgrim,K.F.Willey,D.L.Robbins, and M.A.Duncan,Int.Rev.Phys.Chem.13,231 (1994).
[3]M.A.Duncan,Annu.Rev.Phys.Chem.48,69(1997).
[4]M.A.Duncan,Int.Rev.Phys.Chem.22,407(2003).
[5]N.R.Walker,R.S.Walters,and M.A.Duncan,New J.Chem.29,1495(2005).
[6]J.M.Lisy,Int.Rev.Phys.Chem.16,267(1997).
[7]J.A.Maner,D.T.Mauney,and M.A.Duncan,J.Phys. Chem.Lett.6,449(2015).
[8]H.Ke and J.M.Lisy,Phys.Chem.Chem.Phys.17, 25354(2015).
[9]K.L.Han and G.Z.He,J.Photochem.Photobio.C Photochem.Rev.8,55(2007).
[10]Z.R.Lou,P.Li,and K.L.Han,Acc.Chem.Res.48, 1358(2015).
[11]X.Q.Pang,K.L.Han,and Q.Cui,J.Comput.Chem. 34,1620(2013).
[12]M.A.Duncan,Rev.Sci.Instrum.83,041101(2012).
[13]Y.C.Cheng,J.Chen,L.N.Ding,T.H.Wong,P. D.Kleiber,and D.K.Liu,J.Chem.Phys.104,6452 (1996).
[14]B.Jin,D.S.Wang,and J.Y.Liu,J.Theor.Comput. Chem.10,349(2011).
[15]A.Furuya,H.Tsunoyama,F.Misaizu,and K.Ohno, Chem.Phys.Lett.382,283(2003).
[16]X.Yang,Y.H.Hu,and S.H.Yang,J.Phys.Chem.A 104,8496(2000).
[17]K.F.Willey,C.S.Yeh,D.L.Robbins,J.S.Pilgrim, and M.A.Duncan,J.Chem.Phys.97,8886(1992).
[18]F.Misaizu,M.Sanekata,K.Fuke,and S.Iwata,J. Chem.Phys.100,1161(1994).
[19]M.Sanekata,F.Misaizu,and K.Fuke,J.Chem.Phys. 104,9768(1996).
[20]J.Qian,A.J.Midey,S.G.Donnelly,J.I.Lee,and J. M.Farrar,Chem.Phys.Lett.244,414(1995).
[21]J.I.Lee,D.C.Sperry,and J.M.Farrar,J.Chem.Phys. 114,6180(2001).
[22]W.Y.Lu and P.D.Kleiber,J.Chem.Phys.114,10288 (2001).
[23]W.Y.Lu,T.H.Wong,Y.Sheng,and P.D.Kleiber,J. Chem.Phys.117,6970(2002).
[24]Y.H.Hu,R.Chen,L.Chen,X.J.Wang,H.C.Liu, and S.H.Yang,Chem.Phys.Lett.430,255(2006).
[25]H.C.Liu,Y.H.Hu,S.H.Yang,W.Y.Guo,Q.T.Fu, and L.Wang,J.Phys.Chem.A 110,4389(2006).
[26]S.Yoshida,N.Okai,and K.Fuke,Chem.Phys.Lett. 347,93(2001).
[27]W.Y.Guo,H.C.Liu,and S.H.Yang,J.Chem.Phys. 116,2896(2002).
[28]H.C.Liu,W.U.Guo,and S.J.Yang,J.Chem.Phys. 115,4612(2001).
[29]Y.H.Hu,H.H.Liu,and S.H.Yang,J.Chem.Phys. 120,2759(2004).
[30]J.L.Sun,H.C.Liu,H.M.Yin,K.L.Han,and S.H. Yang,J.Phys.Chem.A 108,3947(2004).
[31]W.Y.Guo,H.C.Liu,and S.H.Yang,J.Chem.Phys. 116,9690(2002).
[32]X.Yang,K.L.Gao,H.C.Liu,and S.H.Yang,J. Chem.Phys.112,10236(2000).
[33]H.C.Liu,C.S.Wang,W.Guo,Y.D.Wu,and S.H. Yang,J.Am.Chem.Soc.124,3794(2002).
[34]H.C.Liu,S.H.Yang,X.H.Zhang,and Y.D.Wu,J. Am.Chem.Soc.125,12351(2003).
[35]H.C.Liu,J.L.Sun,Y.H.Hu,K.L.Han,and S.H. Yang,Chem.Phys.Lett.389,342(2004).
[36]C.T.Scurlock,S.H.Pullins,and M.A.Duncan,J. Chem.Phys.105,3579(1996).
[37]C.T.Scurlock,S.H.Pullins,.J.E.Reddic,and M.A. Duncan,J.Chem.Phys.104,4591(1996).
[38]M.R.France,S.H.Pullins,and M.A.Duncan,J. Chem.Phys.109,884(1998).
[39]S.S.Wesolowski,R.A.King,H.F.Schaefer,and M. A.Duncan,J.Chem.Phys.113,701(2000).
[40]J.Chen,Y.C.Cheng,and P.D.Kleiber,J.Chem. Phys.106,3884(1997).
[41]J.H.Holmes,P.D.Kleiber,D.A.Olsgaard,and K.H. Yang,J.Chem.Phys.112,6583(2000).
[42]W.Y.Lu,T.H.Wong,Y.Sheng,and P.D.Kleiber,J. Chem.Phys.118,6905(2003).
[43]T.H.Wong,C.Freel,P.D.Kleiber,and K.M.Sando, J.Chem.Phys.108,5723(1998).
[44]D.S.Wang,K.L.Han,and S.H.Yang,Acta Phys.-Chim.Sin.21,583(2005).
[45]D.S.Wang,K.L.Han,and S.H.Yang,Chin.J.Chem. Phys.19,123(2006).
[46]H.C.Liu,S.H.Yang,X.H.Zhang,and Y.D.Wu,J. Am.Chem.Soc.125,12351(2003).
[47]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09 Revision D.01, Wallingford,CT:Gaussian Inc.,(2009).
[48]L.N.Ding,M.A.Young,P.D.Kleiber,W.C.Stwalley, and A.M.Lyyra,J.Phys.Chem.97,2181(1993).
(Dated:Received on July 4,2016;Accepted on August 11,2016)
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年5期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年5期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Preparation of Bio-hydrogen and Bio-fuels from Lignocellulosic Biomass Pyrolysis-Oil
- Combination Computing of Support Vector Machine,Support Vector Regression and Molecular Docking for Potential Cytochrome P450 1A2 Inhibitors
- Working Condition Real-Time Monitoring Model of Lithium Ion Batteries Based on Distributed Parameter System and Single Particle Model
- Hydrodeoxygenation of Anisole over Ni/α-Al2O3Catalyst
- Highly Efficient and Selective Removal of Pb(II)ions by Sulfur-Containing Calcium Phosphate Nanoparticles
- Efficient Removal Phenol Red over Ternary Heterostructured Ag-Bi2MoO6/BiPO4Composite Photocatalyst