
Comparative genome analysis between Southeast Asian and South American Zika viruses
2016-11-24 11:34:26TheeraratKochakarnNamfonKotananKrittikornmpornsinDuangkamonLoesbanluechaiMontaThammasattaPrasertAuewarakulPraponWilairatThanatChookajorn
Theerarat Kochakarn, Namfon Kotanan, Krittikorn Kümpornsin, Duangkamon Loesbanluechai, Monta Thammasatta, Prasert Auewarakul, Prapon Wilairat, Thanat Chookajorn✉
1Genomics and Evolutionary Medicine Unit, Centre of Excellence in Malaria Research, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand
2Department of Biochemistry, Faculty of Science, Mahidol University, Bangkok, Thailand
3Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University Bangkok, Thailand
4National Science and Technology Development Agency, Pathum Thani, Thailand
5Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
Comparative genome analysis between Southeast Asian and South American Zika viruses
Theerarat Kochakarn1,2, Namfon Kotanan1, Krittikorn Kümpornsin1, Duangkamon Loesbanluechai3, Monta Thammasatta4, Prasert Auewarakul5, Prapon Wilairat2, Thanat Chookajorn1✉
1Genomics and Evolutionary Medicine Unit, Centre of Excellence in Malaria Research, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand
2Department of Biochemistry, Faculty of Science, Mahidol University, Bangkok, Thailand
3Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University Bangkok, Thailand
4National Science and Technology Development Agency, Pathum Thani, Thailand
5Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
ARTICLE INFO
Article history:
Accepted 25 September 2016
Available online 20 November 2016
Phylogenetic analysis
Protein structure
Southeast Asia
Dengue virus
Zika virus
Objective: To understand the cause for the differences between potentially mild Southeast Asian and the more pathogenic ZIKV in South America. Methods: A comparative genomic analysis was performed to determine putative causations stemming from ZIKV. Results: Phylogenetic analyses integrating geographical and time factors revealed that Southeast Asian ZIKV might not be the direct source of South American outbreaks as previously speculated. Amino acid residues unique to South American ZIKV isolates at the envelope, pr and NS1 proteins are listed and shown in the structural context. These unique residues on external viral proteins are not found in Southeast Asian ZIKV and could be responsible for the ongoing outbreak either via an intrinsic property of the virus or interactions with human immunity. Only a selected few primer/probe sets currently in clinical use were identified of being capable of detecting ZIKV strains worldwide. The envelope proteins of dengue virus (DENV) and ZIKV also showed a remarkable degree of similarity especially at the surface residues. Conclusions: findings that may help explain the cross-reactivity of DENV antibodies to ZIKV. Thus, major caveats must be exercised in using existing diagnostic tools for ZIKV.
1. Introduction
Zika virus (ZIKV), a member of the vector-borne Flaviviridaefamily, has emerged as a new public health threat due to a series of recent outbreaks and links to systemic neurological pathologies [1]. Symptoms of ZIKV infections include fever, rash, conjunctivitis and arthralgia, which can often lead to misdiagnosis as the more common dengue virus (DENV) infection[2]. The spread of ZIKV now has become a critical and urgent issue because the 2015 ZIKV outbreak in northern Brazil coincided with a spike in the incidence of microcephaly in newborns [3]. The infection was also linked to systemic neurological disorders and morphological alterations of neuronal organs [1]. ZIKV has been identified in brain tissue and amniotic fluid from microcephaly cases [4, 5]. ZIKV neurotropism and direct causation were shown both at the cellular level and in an animal model [6-8].
Despite its discovery in Africa, ZIKV has been found in different parts of the world [9]. In Southeast Asia, cases of ZIKV infection have been reported intermittently during the past ten years [10-13]. In Thailand alone, ZIKV infections have been identified throughout the country [14]. However, the infection often is misdiagnosed as DENV infection due to their similar symptoms and cross-reactive immunological antigens, which currently limits our understanding of ZIKV pathology [14]. Interestingly, despite the long period of ZIKV circulation in Thailand and Southeast Asia, there is no direct evidence linking this virus in the region to any neurological disorder. The difference in neurotropism might be the result of ZIKV genetic variations between the two continents. Nevertheless, it is not possible to disregard the possibility that the neurological damage might be the result of host-virus interactions specific to South America. In addition, the number of ZIKV cases per area in Southeast Asia might not be sufficiently high to manifest ZIKV-related neurological symptoms. In order to introduce a well-informed strategy to combat ZIKV infection, it is important to urgently address the aforementioned issues. Thorough comparative analyses of available ZIKV genomes from various geographical origins will be highly informative in providing clues towards determining and containing the threat of ZIKV infection.
In this study, genetic compositions of ZIKV protein motifs were analyzed to identify possible unique features from viruses responsible for the South America outbreak and not found in those circulating in Southeast Asia and Africa. Comparative genomic analyses also allowed us to determine the quality of ZIKV diagnostic methods employed to detect ZIKV isolates from different countries. Comparative analysis with four DENV subtypes revealed the underlying causes for the cross-reactivity of anti-DENV antibodies to ZIKV, which could compromise immunological detection approaches.
2. Materials and methods
2.1. Genomic sequence analysis
Thirty-three ZIKV genomic sequences were retrieved from NCBI Nucleotide database [4, 5, 11, 15-22]. The entries were named according to their geographical origins and periods of collection as follows: BrazilAmniotic2015 (KU497555), BrazilBrain2015 (KU527068), Brazil2015_1 (KU365777), Brazil2015_2 (KU365778), Brazil2015_3 (KU365779), Brazil2015_4 (KU365780), Brazil2015_5 (KU707826), Cambodia2010 (JN860885), CAR1968 (KF383115), CAR1976 (KF268950), CAR1979 (KF268948), CAR1980 (KF268949), FrenchPolynesia2013 (KJ776791), Guatemala2015_1 (KU501216), Guatemala2015_2 (KU501217), Haiti2014 (KU509998), Malaysia1966 (HQ234499), Martinique2015 (KU647676), Nigeria1968 (HQ234500), Philippines2012 (KU681082), PuertoRico2015 (KU501215), SaoPaulo2015 (KU321639), Senegal1968 (KF383116), Senegal1984 (HQ234501), Senegal1997 (KF383117), Senegal2001 (KF383119), Suriname2015 (KU312312), Thailand2013 (KF993678), Thailand2014 (KU681081), Uganda1947_1 (AY632535), Uganda1947_2 (HQ234498), Uganda1947_3 (LC002520) and Yap2007 (EU545988). Alignment of genomic sequences was performed with MAFFT and visualized using Jalview version 2.8 [23, 24]. Neighbor-joining (NJ) tree and 1 000 bootstrap replicates were calculated using ClustalX version 2.0 [25]. Maximum likelihood (ML) tree and 1 000 bootstrap replicates were estimated using GTR+ γ+I substitution model and RAxML version 8.2.8 [26]. Bayesian analysis was performed with BEAST version 1.8.3, using a strict molecular clock in the GTR+γ+I substitution model [27]. Diagrams of phylogenetic trees were constructed using Dendroscope version 3.5.7 [28]. Polyprotein sequences were decoded from genomes by EMBOSS Transeq with amino acid variations listed by Jalview. Gene annotation was based on Uganda1947_1, and transmembrane helices were predicted using TMHMM Server v. 2.0 [16, 29].
2.2. Homology model construction
SWISS-MODEL server was employed for template search and initial model building [30]. Models were refined by KoBaMIN and checked for Ramachandran outlier by RAMPAGE [31, 32]. Templates used for model constructions are described in the text below. Protein structures with root-mean-square deviation (RMSD) were visualized and calculated using PyMOL version 1.3.
In order to compare ZIKV envelope protein with that of DENV, MAFFT was used to align the envelope protein sequence from ZIKV BrazilBrain2015 isolate with the sequences of envelope protein from DENV type 1 (NCBI accession number NC_001477), type 2 (TB16i strain, NCBI accession number AY858036.2), type 3 (TB55i strain, NCBI accession number AY858048.2), and type 4 (DENV-4/KH/ BID-V2055/2002 isolate, NCBI accession number KF955510.1). The conservation score was calculated based on blocks substitution matrix BLOSUM62.
3. Results
3.1. Phylogenetic branching of Southeast Asian-South American ZIKV descents
Of the 33 ZIKV genomes from various sources employed in this study, five originated in Southeast Asia. Structures of phylogenetic trees of the whole genome constructed using ML, NJ and Bayesian methods are not significantly different, with ZIKV isolates from Africa grouping together apart from ZIKV isolates from Asia, Pacific Islands and South America (Figure 1A). A large portion of the genomic sequences was from Africa samples collected several decades ago, and they clustered together as a separate branch. The first available Southeast Asia ZIKV data was from the Malaysian sample collected in 1966. This strain appears as an outgroup in the phylogenetic tree. When focus was placed on comparing Southeast Asian with South America ZIKV isolates, the latter viruses were more closely related to ZIKV from French Polynesia of 2013 than to Southeast Asian isolates (Figures 1A and B). South American ZIKV might share ancestors with Southeast Asian ZIKV, but they appear not to be a direct descendent. Even though ‘recent' ZIKV has circulated in the Southeast Asia region since 2010, there was no reports on ZIKV outbreaks or ZIKV-related microcephaly cases as in South America. It is not yet known whether the recent large-scale outbreaks and microcephaly were caused by a specific functionalgain caused by novel mutations. Understanding the significance of the specific changes in the ZIKV proteins should provide the first set of clues toward an understanding of the genetics underlying the difference between potentially mild Southeast Asian and more virulent South American ZIKV strains.
3.2. Potential selective signatures of ZIKV genomes and proteins
Comparison of ZIKV genomes ought to expose any genetic footprints left by evolutionary pressure in the form of positive selection or reshuffled tree structures. Analysis of ZIKV genome organization revealed an overall genome structure similar to that of DENV. The 10.7 kb ZIKV single-stranded RNA genome encodes a single polypeptide chain of 3,419 amino acid residues with noncoding 5´- and 3´-UTRs (Figure 1C). This ZIKV polypeptide chain is likely to be inserted into the endoplasmic reticulum membrane via a translational/translocation-coupled process and be cleaved into individual polypeptide fragments consisting of three structural proteins that form the viral particle (C, prM and E) and seven nonstructural proteins that are involved in viral propagation (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5). Analysis of the phylogenetic pattern of each individual ZIKV protein showed similar tree structures consistent with analysis performed using the whole genome [33]. The dN/dS ratio of ZIKV is relatively low for every ZIKV proteins, indicating that the proteins are not under positive selection, even for the surface proteins [33].
In order to explore the presence of possible selective signatures, comparative analysis was conducted using protein structures, which could reveal selections at specific residues, functional domains or catalytic sites. Differences between ZIKV isolates in Southeast Asia circulation and those from South American outbreaks were analyzed by noting amino acid substitutions that are uniquely found in South American and not Southeast Asian and African ZIKV isolates, with particular focus on virus proteins that are externally exposed, namely, envelope, pr and NS1, since they are likely to interact with host factors mediating host immune responses. Because information on ZIKV protein structures is available only for envelope protein, membrane protein and NS1β-ladder, models of other ZIKV proteins were generated based on the closest available structures from flavivirus proteins [34-39].
In the case of ZIKV NS1, we have generated a homology model of the full length protein. The model has C RMSD of 0.742 Å from the C-αatoms of ZIKV NS1β-ladder domain (PDB ID: 5IY3) [36]. In DENV, NS1 protein is required for virus propagation and also acts as a virulent factor [40]. DENV NS1 protein is secreted from infected cells in the form of a hexameric complex with a central hydrophobic channel [38, 41, 42]. Five amino acid residues specific to South American ZIKV isolates are on the surface of this protein (Figure 2A). The planar side facing outward from the South American NS1 hexamer has A100, A233 and V349 compared with Southeast Asian and African isolates (G100, T233 and M349). A233 and V349 were found in Brazilian ZIKV isolate from the brain of a microcephalic fetus [4]. There are also two South American specific substitutions at the inner lining of ZIKV NS1 hexamer center, namely, H122 and W324 (compared with Y122 and R324 in Southeast Asian and African isolates).of immature virus particles within the trans-Golgi.

Figure 1. Phylogenetic analysis of Southeast Asian and South American ZIKV branches. A) Bayesian phylogenetic tree of ZIKV open reading frames from 33 isolates are shown with samples from Africa (yellow), Southeast Asia (green), Pacific Islands (cyan) and South America (orange). The horizontal gray bars at each node represents 95% Bayesian confidence intervals for divergence periods. Numbers at the nodes represent posterior probabilities. To separate new and old samples, isolates collected within ten years are labeled with white diamonds. The ones collected earlier than that are marked with black diamonds. B) Bayesian phylogenetic tree of with the focus on the isolates collected during the last ten years. When the old samples were removed, the separation between the South American and Southeast Asian groups becomes more noticeable. The color codes and labels are similar to those in Fig. 1A. Numbers at the nodes represent posterior probabilities. C) Diagram of ZIKV genome and putative organization were constructed in comparison to those of DENV. The long ZIKV polypeptide chain is likely to be inserted into membrane, allowing further processing in the ER-golgi complex. The upper panel represents putative RNA secondary structures predicted by Mfold and a putative protein diagram. The lower panel shows the arrangement of proteins with matching colors.
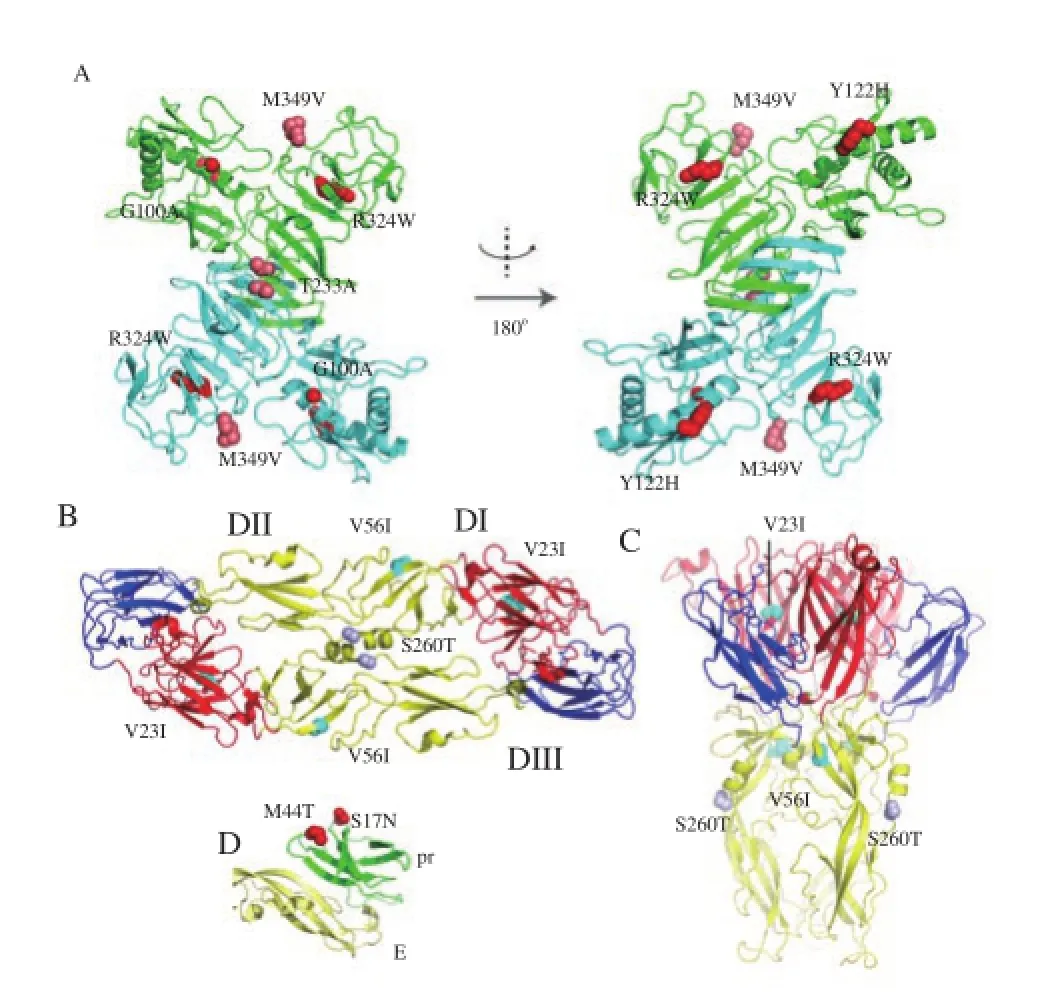
Figure 2. Amino acid residues of the envelope, pr peptide and NS1 proteins that are unique in the South American ZIKV isolates.
The mature ZIKV particle has 180 copies of envelope protein in the form of homodimers [34, 35]. This protein mediates host cell receptor binding and is recognized by host antibodies [43-45]. During host cell invasion, the envelope protein forms a homotrimer that functions to facilitate fusion of virus envelope with the host endosome membrane[46]. There are three amino acid substitutions in the envelope ectodomain that are unique to South Amearica ZIKV: T260, exposed in both dimer and trimer, and present exclusively in Brazilian ZIKV isolated from amniotic fluid of fetus with microcephaly; I23 and I56, located at the hydrophobic domain (Figure 2B and C).
The prM protein functions as a protector against premature ZIKV fusion with host cell membrane by binding to the fusion loop domain of the envelope protein [47-49]. PrM on the immature virus particle is cleaved to pr peptide and membrane protein within the trans-Golgi [48, 49]. Two substitutions located on the opposite side to the fusion loop are present in South American ZIKV: N17, found in all South American ZIKV but also in an isolate from French Polynesia collected in 2013; and T44, identified in Suriname (Figure 2D). These two residues are on the pr peptide and present on the surface
3.3. Genome data analysis and implications for current diagnostic tools
In Thailand, DENV is endemic with more than a hundred thousand cases every year [50]. ZIKV is overlooked in the country as its clinical presentations and managements are the same as DENV cases. In addition, the commonly used serological methods for DENV diagnosis falsely detect ZIKV as DENV [14]. Molecular diagnostics of ZIKV relies on reverse-transcriptase polymerase chain reaction (RT-PCR) and TaqMan probes [2]. However, the probes employed in such studies tend to rely on sequence information available at the time. It was not possible then to determine whether the probe and primer sets can universally identify ZIKV. Comparative genomic data of ZIKV could provide data regarding the quality of each ZIKV detection primer and probe sets.
When the probe and primer sets commonly used in ZIKV diagnosis worldwide were matched with genome data, it becomes alarmingly clear that some of them would fail to detect a significant portion of ZIKV strains. To address this pressing issue, a collection of primer sets is recommended here for their versatility in detecting all or almost all ZIKV strains (Table 1). The best design uses a degenerate primer/probe set targeting NS5 that perfectly matches every strain with only two exceptions of ZIKV from the Central African Republic [51]. The TaqMan probe set designed by the Pan American Health Organization also recognizes strains currently in circulation with a few mismatches of African ZIKV strains collected some decades back [2]. Information regarding published primer/probe sets for ZIKV detection can be found at http://www.tm.mahidol.ac.th/ gem/sites/default/files/pictures/ZikaPrimersAlignment.pdf [2, 15, 51-55].

Table 1 Recommended probe/primer sets for ZIKV detection by RT-PCR.
An alternative method for detecting ZIKV is the use of immunobased methods. The key epitopes for antibody detection are likely to be located on the ectodomain of the envelope protein. Comparisons between the envelope proteins of ZIKV and DENV to identify epitope regions unique to each virus species showed that the ZIKV envelope protein shares a high degree of similarity with those of the four DENV subtypes (Figure 3A). Remarkably, structuralcomparison identified matching residues that are located at the external surface (Figure 3B). Matching residues occupy a large portion of the protein surface, and only a region near the glycosylation site in the DI domain (residues 157-168, 178-179 and 181-182), a small loop in the DII domain (residues 277-283) and a small loop in the DIII domain (residues 364-370) present potential epitope sites sufficiently dissimilar to distinguish between the two virus groups.
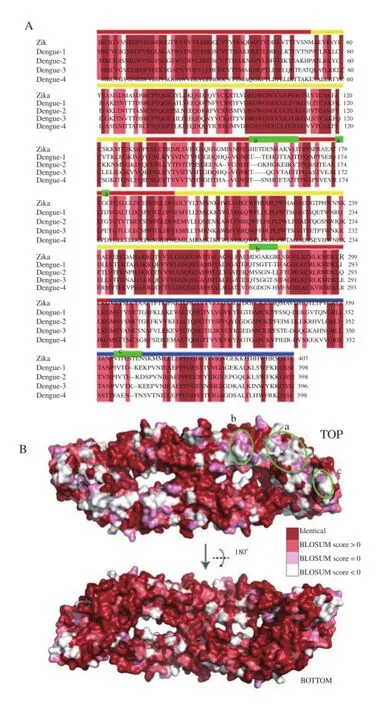
Figure 3. Similarity between the envelope proteins of DENV and ZIKV.
4. Discussion
Here we present a comparative analysis of ZIKV focusing on the differences between Southeast Asian and South American ZIKV populations. The incentive for the study was the research question as to why ZIKV populations in Southeast Asia do not cause outbreaks and microcephaly on a large scale. The issue is pressing because the upcoming 2016 Summer Olympics in Brazil might globally cause population admixtures of ZIKV. We compared ZIKV from Southeast Asia and South America in the context of available structural information. The amino acid residues externally exposed that are unique to South American ZIKV are identified to highlight their potential as candidates that might be involved in pathogenesis and virus reproductive success. They may also represent sites that can be exploited to differentiate viruses from specific geographical regions. However, functional testing is definitely required to test the significance of each geographic-specific residue. Considering the urgent threat of ZIKV outbreaks, we present here primary data analysis, which we hope will be of immediate benefit to the research and medical communities.
Comparative genomic data also demonstrated the potential and limitation of the primer/probe sets currently in clinical use. Certain sets would fail to detect a large portion of the ZIKV populations. We hope that public health authorities take our observations into consideration and develop ZIKV detection protocols suitable for each geographical region. In addition, the strong degree of similarity between DENV and ZIKV envelope proteins suggests a potential of using dengue antibodies against ZIKV. Antibodies targeting shared epitopes could thwart neurological damages in infected pregnant women. Several dengue vaccines are already in clinical trials with existing safety data [56]. Immunological cross-reactivity between ZIKV and DENV could rapidly be exploited from being a problem into becoming a solution in treating vulnerable populations. The similarity between the two viruses also poses another challenge regarding DENV and ZIKV co-infection. Co-infection cases have already been reported, and it might be just a tip of the iceberg considering their shared geographical areas of endemicity [57]. Differences in degree of severity of DENV infections have been proposed to be the outcome of sequential subtype infections [58, 59]. With the spread of ZIKV, it is necessary now to determine how coinfection and/or sequential infection of ZIKV and DENV influence clinical outcomes.
At present, the number of available ZIKV genomes are still limited. Analysis presented here was conducted to provide the first set of evidence on the strengths and weaknesses of the ZIKV genomic analysis. It is crucial to obtain more genome sequences from diverse geographical areas with accompanying clinical data. A common nomenclature system has been proposed in order to encourage data sharing [60]. Cooperation on ZIKV research is inevitable given that the virus can be found in different parts of the world, each with unique population structures and clinical manifestations. Potential devastating effects of ZIKV pandemics will require the global community to work together to prevent such a scenario.
Declare of interest statement
We declare that we have no conflict of interest.
Acknowledgments
We thank A. Sabchareon, O. Miotto, C. Modchang and P. Ponsuwanna for their comments and suggestions on Zika virus research. The work in the Chookajorn laboratory is supported by Thailand Research Fund-Mahidol University (RSA5880062) and by the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative. TK is supported by the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0204/2552).
References
[1] Brito C. Zika virus: A new chapter in the history of medicine. Acta Med Port 2015;28(6): 679-680.
[2] Waggoner JJ, Pinsky BA. Zika virus: Diagnostics for an emerging pandemic threat. J Clin Microbiol 2016;54(4):860-867.
[3] Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol 2016;47(1): 6-7.
[4] Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, et al. Zika virus associated with microcephaly. N Engl J Med 2016;374(10): 951-958.
[5] Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 2016;16(6): 653-660.
[6] Rubin EJ, Greene MF, Baden LR. Zika virus and microcephaly. N Engl J Med 2016;374(10): 984-985.
[7] Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016;352(6287): 816-818.
[8] Shan C, Xie X, Muruato AE, Rossi SL, Roundy CM, Azar SR, et al. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 2016;19:891-900.
[9] Ioos S, Mallet HP, Leparc Goffart I, Gauthier V, Cardoso T, Herida M. Current Zika virus epidemiology and recent epidemics. Med Mal Infect 2014;44(7): 302-307.
[10] Alera MT, Hermann L, Tac-An IA, Klungthong C, Rutvisuttinunt W, Manasatienkij W, et al. Zika virus infection, Philippines, 2012. Emerg Infect Dis 2015;21(4): 722-724.
[11] Fonseca K, Meatherall B, Zarra D, Drebot M, MacDonald J, Pabbaraju K, et al. First case of Zika virus infection in a returning Canadian traveler. Am J Trop Med Hyg 2014;91(5): 1035-1038.
[12] Marchette NJ, Garcia R, Rudnick A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am J Trop Med Hyg 1969;18(3): 411-415.
[13] Tappe D, Nachtigall S, Kapaun A, Schnitzler P, Gunther S, Schmidt-Chanasit J. Acute Zika virus infection after travel to Malaysian Borneo, September 2014. Emerg Infect Dis 2015;21(5): 911-913.
[14] Buathong R, Hermann L, Thaisomboonsuk B, Rutvisuttinunt W, Klungthong C, Chinnawirotpisan P, et al. Detection of Zika virus infection in Thailand, 2012-2014. Am J Trop Med Hyg 2015;93(2): 380-383.
[15] Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 2008;14(8): 1232-1239.
[16] Kuno G, Chang GJ. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 2007;152(4): 687-696.
[17] Enfissi A, Codrington J, Roosblad J, Kazanji M, Rousset D. Zika virus genome from the Americas. Lancet 2016; 387(10015): 227-228.
[18] Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, et al. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 2012;6(2): e1477.
[19] Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis 2014;8(1): e2636.
[20] Berthet N, Nakoune E, Kamgang B, Selekon B, Descorps-Declere S, Gessain A, et al. Molecular characterization of three Zika flaviviruses obtained from sylvatic mosquitoes in the Central African Republic. Vector Borne Zoonotic Dis 2014;14(12): 862-865.
[21] Baronti C, Piorkowski G, Charrel RN, Boubis L, Leparc-Goffart I, de Lamballerie X. Complete coding sequence of zika virus from a French polynesia outbreak in 2013. Genome Announc 2014;2(3):e00500-14.
[22] Faria NR, Azevedo Rdo S, Kraemer MU, Souza R, Cunha MS, Hill SC, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016;352(6283): 345-349.
[23] Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30(4): 772-780.
[24] Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25(9): 1189-1191.
[25] Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23(21): 2947-2948.
[26] Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30(9): 1312-1313.
[27] Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012;29(8): 1969-1973.
[28] Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 2007;8(1):460.
[29] Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predictingtransmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001;305(3): 567-580.
[30] Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42(Web Server issue): W252-8.
[31] Rodrigues JP, Levitt M, Chopra G. KoBaMIN: a knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res 2012;40(Web Server issue): W323-8.
[32] Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 2003;50(3): 437-450.
[33] Zhu Z, Chan JF, Tee KM, Choi GK, Lau SK, Woo PC, et al. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg Microbes Infect 2016;5:e22.
[34] Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, et al. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016;352(6284): 467-470.
[35] Kostyuchenko VA, Lim EX, Zhang S, Fibriansah G, Ng TS, Ooi JS, et al. Structure of the thermally stable Zika virus. Nature 2016;533(7603): 425-428.
[36] Song H, Qi J, Haywood J, Shi Y, Gao GF. Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat Struct Mol Biol 2016;23(5): 456-458.
[37] Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, et al. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 2008;319(5871): 1830-1834.
[38] Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014;343(6173): 881-885.
[39] Luca VC, Nelson CA, Fremont DH. Structure of the St. Louis encephalitis virus postfusion envelope trimer. J Virol 2013;87(2): 818-828.
[40] Akey DL, Brown WC, Jose J, Kuhn RJ, Smith JL. Structure-guided insights on the role of NS1 in flavivirus infection. Bioessays 2015;37(5): 489-494.
[41] Flamand M, Megret F, Mathieu M, Lepault J, Rey FA, Deubel V. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J Virol 1999;73(7): 6104-6110.
[42] Gutsche I, Coulibaly F, Voss JE, Salmon J, d'Alayer J, Ermonval M, et al. Secreted dengue virus nonstructural protein NS1 is an atypical barrelshaped high-density lipoprotein. Proc Natl Acad Sci U S A 2011;108(19): 8003-8008.
[43] Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol 2005;79(2): 1223-1231.
[44] Lee JW, Chu JJ, Ng ML. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J Biol Chem 2006;281(3): 1352-1360.
[45] Hung SL, Lee PL, Chen HW, Chen LK, Kao CL, King CC. Analysis of the steps involved in Dengue virus entry into host cells. Virology 1999;257(1): 156-167.
[46] Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004;427(6972): 313-319.
[47] Zhang Q, Hunke C, Yau YH, Seow V, Lee S, Tanner LB, et al. The stem region of premembrane protein plays an important role in the virus surface protein rearrangement during dengue maturation. J Biol Chem 2012;287(48): 40525-40534.
[48] Yu IM, Holdaway HA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol 2009;83(23): 12101-12107.
[49] Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, et al. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008;319(5871): 1834-1837.
[50] Corbel V, Nosten F, Thanispong K, Luxemburger C, Kongmee M, Chareonviriyaphap T. Challenges and prospects for dengue and malaria control in Thailand, Southeast Asia. Trends Parasitol 2013;29(12): 623-633.
[51] Faye O, Faye O, Diallo D, Diallo M, Weidmann M, Sall AA. Quantitative real-time PCR detection of Zika virus and evaluation with field-caught mosquitoes. Virol J 2013;10:311.
[52] Faye O, Faye O, Dupressoir A, Weidmann M, Ndiaye M and Alpha Sall A. One-step RT-PCR for detection of Zika virus. J Clin Virol 2008;43(1): 96-101.
[53] Tappe D, Rissland J, Gabriel M, Emmerich P, Gunther S, Held G, et al. First case of laboratory-confirmed Zika virus infection imported into Europe, November 2013. Euro Surveill 2014;19(4):12-15.
[54] Balm MN, Lee CK, Lee HK, Chiu L, Koay ES, Tang JW. A diagnostic polymerase chain reaction assay for Zika virus. J Med Virol 2012;84(9): 1501-1505.
[55] Pyke AT, Daly MT, Cameron JN, Moore PR, Taylor CT, Hewitson GR, et al. Imported zika virus infection from the cook islands into australia, 2014. PLoS Curr 2014;6:DOI:10.1371/currents.outbreaks.4635a54dbffba 2156fb2fd76dc49f65e.
[56] Screaton G, Mongkolsapaya J, Yacoub S, Roberts C. New insights into the immunopathology and control of dengue virus infection. Nat Rev Immunol 2015;15(12): 745-759.
[57] Dupont-Rouzeyrol M, O'Connor O, Calvez E, Daures M, John M, Grangeon JP, et al. Co-infection with Zika and dengue viruses in 2 patients, New Caledonia, 2014. Emerg Infect Dis 2015;21(2): 381-382.
[58] Mongkolsapaya J, Dejnirattisai W, Xu XN, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med 2003;9(7): 921-927.
[59] Halstead SB. Controversies in dengue pathogenesis. Paediatr Int Child Health 2012;32(Suppl):15-19.
[60] Scheuermann RH. Zika virus: designate standardized names. Nature 2016;531(7593): 173.
Document heading 10.1016/j.apjtm.2016.10.002
28 July 2016
in revised form 20 September 2016
Theerarat Kochakarn, Genomics and Evolutionary Medicine Unit, Centre of Excellence in Malaria, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand.
✉ Thanat Chookajorn, Genomics and Evolutionary Medicine Unit, Centre of Excellence in Malaria Research, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand.
E-mail: thanat.cho@mahidol.edu
Foundation project: This work in the Chookajorn laboratory is supported by Thailand Research Fund-Mahidol University (RSA5880062) and by the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative. TK is supported by the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0204/2552).
 Asian Pacific Journal of Tropical Medicine2016年11期
Asian Pacific Journal of Tropical Medicine2016年11期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Modifiable determinants of attitude towards dengue vaccination among healthy inhabitants of Aceh, Indonesia: Findings from a communitybased survey
- Clinical significance of dynamic detection for serum levels of MCP-1, TNF-α and IL-8 in patients with acute pancreatitis
- Expression and mechanism of action of miR-196a in epithelial ovarian cancer
- Protective effect of antioxidant on renal damage caused by Doxorubicin chemotherapy in mice with hepatic cancer
- Mechanism of action of Zhuyu Annao pill in mice with cerebral intrahemorrhage based on TLR4
- Acetylcholinesterase, butyrylcholinesterase and paraoxonase 1 activities in rats treated with cannabis, tramadol or both