
Iron dysregulation in beta-thalassemia
2016-11-24 11:34:25KamonlakLeecharoenkiatPathrapolLithanatudomWannapaSornjaiDuncanSmith
Kamonlak Leecharoenkiat, Pathrapol Lithanatudom, Wannapa Sornjai, Duncan R. Smith✉
1Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, 154 Rama 4 Road, Bangkok, Thailand 10330
2Department of Biology, Faculty of Science, Chiang Mai University, 239 Huaykaew Road, Amphur Muang, Chiang Mai, Thailand 50200
3Molecular Pathology Laboratory, Institute of Molecular Biosciences, Mahidol University, 25/25 Phuttamonthon 4 Road, Salaya, Nakhon Pathom, Thailand 73170
Iron dysregulation in beta-thalassemia
Kamonlak Leecharoenkiat1, Pathrapol Lithanatudom2, Wannapa Sornjai3, Duncan R. Smith3✉
1Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, 154 Rama 4 Road, Bangkok, Thailand 10330
2Department of Biology, Faculty of Science, Chiang Mai University, 239 Huaykaew Road, Amphur Muang, Chiang Mai, Thailand 50200
3Molecular Pathology Laboratory, Institute of Molecular Biosciences, Mahidol University, 25/25 Phuttamonthon 4 Road, Salaya, Nakhon Pathom, Thailand 73170
ARTICLE INFO
Article history:
Accepted 1 August 2016
Available online 20 November 2016
Beta-thalassemia
Hepcidin
Iron overload
Ferroportin
Iron deficiency anemia and iron overload conditions affect more than one billion people worldwide. Iron homeostasis involves the regulation of cells that export iron into the plasma and cells that utilize or store iron. The cellular iron balance in humans is primarily mediated by the hepcidin-ferroportin axis. Ferroportin is the sole cellular iron export protein, and its expression is regulated transcriptionally, post-transcriptionally and posttranslationally. Hepcidin, a hormone produced by liver cells, post-translationally regulates ferroportin expression on iron exporting cells by binding with ferroportin and promoting its internalization by endocytosis and subsequent degradation by lysosomes. Dysregulation of iron homeostasis leading to iron deposition in vital organs is the main cause of death in betathalassemia patients. Beta-thalassemia patients show marked hepcidin suppression, ineffective erythropoiesis, anemia and iron overload. Beta-thalassemia is common in the Mediterranean region, Southeast Asia and the Indian subcontinent, and the focus of this review is to provide an update on the factors mediating hepcidin related iron dysregulation in beta-thalassemia disease. Understanding this process may pave the way for new treatments to ameliorate iron overloading and improve the long term prognosis of these patients.
1. Introduction
With the exception of a few species of bacteria, all living things need iron as an absolute requirement for viability. The ability of iron to act as both an electron donor and an electron acceptor makes it a critical component of many cellular oxidation/reduction reactions,and in addition iron is the substrate for heme, the critical component of hemoglobin, the essential oxygen carrying molecule of all vertebrates [1]. However, free iron is potentially extremely toxic to cells. Iron can donate electrons to oxygen resulting in the formation of the reactive superoxide radical (O2-) or to hydrogen peroxide generating the hydroxyl (·OH) radical [1], and these molecules can oxidize biological macromolecules including lipids, proteins and DNA with extremely damaging consequences to the cell [2].
Humans contain approximately 3 g -4 g of iron in various forms [3]. Although iron is extremely plentiful in the environment, much of it is present in insoluble, non-bioavailable forms, and so humans have evolved to be highly efficient in conserving iron. Indeed, humans have no mechanism for excretion of excess iron under conditions of iron overload. Bioavailable iron in the diet serves mainly to replace iron lost from the body through processes such as the shedding of cells from the surface of the skin and lumen of the gut as part of the normal process of epithelial cell turnover. Additional loss of iron from the body may occur through minor bleeding events. In general it is believed that only some 1 mg to 2 mg of iron (less than 0.1% of the totaliron in a body) are lost from the body each day that require replacement though dietary sources [3].
The majority of the iron in humans is in the form of hemoglobin in red blood cells, and red blood cells combined contain between 2 g and 2.5 g of iron (out of 3 g to 4 g in total). Red blood cells have a life span of some 120 days under normal conditions [2], after which they are degraded by macrophages and the iron returned to the plasma. Plasma contains some 2 mg-3 mg of iron bound to a protein called transferrin, which is the primary molecule that transports iron for use in erythropoiesis in the bone marrow and by other iron requiring cells. Macrophages return some 20 mg -25 mg of iron daily, ensuring a rapid turnover of iron in the plasma. When the binding capacity of serum transferrin is exceeded, iron starts to make complexes with other plasma proteins and molecules such as citrate. This iron is generally termed non-transferrin bound iron (NTBI) [4]. NTBI is easily taken up by hepatocytes and other parenchymal cells, and the intracellular accumulation of iron in these cells rapidly causes damage through oxidation reactions [2].
Within cells, iron is normally stored as the ferric (Fe3+form) in association with a globular protein complex called ferritin. Ferritin is essentially a hollow sphere in which can sequester up to 4 500 iron atoms. The ferritin complex consists of 24 subunits of heavy (H) and light (L) chains the exact composition of which can vary between tissues. The H chain has ferroxidase activity which converts Fe2+to Fe3+for storage inside the shell, while the L chain primarily stabilizes the structure and facilitates transport of iron ions to the inside of the structure [1]. This formation is the main storage system of iron (outside of the iron in hemoglobin). Under conditions of iron deficiency, iron is released from the complex to the plasma, while under conditions of mild iron excess the system can provide some buffering against the increased iron levels. In the average male, one gram of iron is held in storage mostly in hepatocytes and macrophages in the liver but also in spleen red pulp macrophages. Women of reproductive age tend to have significantly lower stored iron as a consequence of menstruation and childbearing [5].
As noted above, only a very small fraction of the total iron content is lost daily, and this is replaced through bioavailable iron sourced from the diet. Iron from the diet can be obtained from heme based sources (found in meat) and from non-heme iron sources (iron in cereals, vegetables, pulses etc). Iron absorption takes place in the gut duodenum and upper jejunum and occurs by transport across the apical membrane of enterocytes, which appears to occur through two independent pathways [6], one for heme iron and one for non-heme iron [3]. While absorption of non-heme iron is fairly well understood, the absorption of heme iron and ferritin iron is rather less well understood. Dietary non-heme iron is normally in the form of ferric iron (Fe3+) which is reduced in the gut to the ferrous (Fe2+) form by ferric reductase activity provided by duodenal cytochrome B and possibly Steap2 [3]. The ferrous iron is then transported across the apical (gut lumen) side of enterocytes by the ferrous iron transporter divalent metal ion transporter 1 (DMT-1), also known as Nramp-2 (natural resistance-associated macrophage protein) [7]. Some evidence suggests that heme iron may be taken up by receptor mediated endocytosis, although no high-affinity heme receptor has been identified to date [3]. There is some evidence that dietary ferritin is also taken up by endocytosis [8]. Once inside the enterocyte heme is broken down by heme oxygenase and dietary ferritin iron is released from ferritin. It is currently believed that iron from the various sources enters a common iron pool within the enterocyte. Some of the iron may be stored directly within the enterocyte as ferritin, while other iron will be released from the cell to end up bound to blood transferrin [8].
2. Intracellular iron trafficking and transportation
There is only one known cellular iron exporter, namely ferroportin [9-11]. This protein is found on the basolateral membrane of enterocytes as well as on other cells such as reticuloendothelial macrophages that export recycled iron, hepatocytes that release storage iron and on differentiating erythrocytes. Ferroportin exports iron in the ferrous (Fe2+) form, but transferrin binds iron in the ferric (Fe3+) form, so ferroxidases are believed to play a role in iron export. In intestinal enterocytes it is believed that hephaestin is the active ferroxidase, while in other cells this action is performed by the either circulating or GPI-linked multicopper ferroxidase ceruloplasmin [3, 5]. Once bound to transferrin the iron is delivered to peripheral tissues by the transferrin-transferrin receptor system. After binding to the transferrin receptor, transferrin is internalized by receptor mediated endocytosis and upon acidification of the endosome iron is released from transferrin and converted to the ferrous form (Fe2+) by the ferrireductase Steap family proteins [12, 13] and transported across the membrane of the endosome into the cytoplasm by the action of the ferrous iron transporter divalent metal ion transporter 1 (DMT-1) protein [3, 5].
Ferroportin has been shown to be regulated transcriptionally in enterocytes and macrophages [11, 14] and to be translationally regulated by the iron responsive element (IRE) present in the 5'-UTR of the ferroportin mRNA through the action of iron regulatory proteins (IRP). The IRE-IRPs system is controlled by intracellular iron levels [9, 11, 15, 16]. IRPs are activated during low iron condition under which they bind to the IRE of ferroportin mRNA resulting in translational suppression. Restrained ferroportin expression leads to reduced iron export, maintaining iron for cellular requirements. In addition, ferroportin is regulated at a posttranslational step by the master iron homeostasis hormone, hepcidin. In erythroid precursor cells (and in enterocytes) a second mRNA encoding for ferroportin has been reported [17, 18]. This mRNA is produced by the use of an alternate, upstream gene promoter and has an identical open reading frame in the mRNA, and as such the protein produced is identical. Critically, this second mRNA (termed FPN1B) does not contain an IRE in the 5'-UTR, and as such is not regulated by iron deficit [18]. It is currently believed that during erythropoiesis the relative expression of these two messages is coordinated to ensure that iron is exported from the cells during early differentiation, but kept in the cells during late differentiation when heme synthesis begins and iron demand is at its highest [18].
2.1. Iron regulation by hepcidin
The absorption of iron by enterocytes, the efflux of recycled iron from macrophages and the efflux of stored iron by hepatocytes are all systemically controlled by the 25 amino-acid peptide hormone hepcidin [19, 20] which is produced predominantly in hepatocytes.Hepcidin is initially synthesized as an 84 amino acid preprohormone, before undergoing processing to generate a 60 amino acid prohormone and finally a 25 amino acid hormone [19]. The structure of the mature hormone is a compactly folded protein with 32% beta-sheet and 4 disulphide bonds [19]. Hepcidin regulates iron efflux by post-translationally negatively regulating ferroportin, the sole iron efflux channel. When iron is present in the plasma in excess hepcidin is secreted from the liver into the plasma. Hepcidin then directly binds to ferroportin expressed on the surface of iron storage cells, triggering endocytosis of both ferroportin and hepcidin which are subsequently degraded by lysosomes [21]. A reduction of ferroportin expression on the cell surface results in less intracellular iron being exported from storage cells, effectively locking iron inside the cell. This event reduces iron efflux into the plasma, returning iron to regular levels. The mechanism by which hepcidin regulates absorption of dietary iron is less clear as ferroportin is located on the basal surface of enterocytes cells, while absorption of dietary iron occurs on the apical surface. The mechanism by which a reduction in the basolateral ferroportin is communicated to the apical iron absorption mechanism remains unclear, although rising intracellular iron levels (as a consequence of reduced efflux) may play a role [5, 21]. Mechanistically however it is known that hepcidin inhibits the uptake step of duodenal iron absorption but does not affect the proportion of iron transferred to the circulation [22].
2.2. Regulation of hepcidin expression
Hepcidin is primarily produced by hepatocytes [21], and hepcidin production is regulated by a number of factors, although it is believed that the primary stimuli is the level of iron in the plasma. Some studies have suggested that the two transferrin receptors (TfR1 and TfR2) together with the membrane protein hereditary hemochromatosis protein (HFE) serve to sense iron levels in the body and induce hepcidin expression, but the mechanism is incompletely understood [5]. A second pathway, the bone morphogenic protein (BMP) pathway, is probably activated in response to iron levels in intracellular iron stores, which results in increased expression of BMP6 which binds to a BMP receptor (BMPR) in association with a co-receptor hemojuvelin which activates the Smad signaling pathway resulting in increased hepcidin expression [5]. Recent evidence has shown that TMPRSS6, which encodes a hepatocytespecific type II transmembrane serine protease, matriptase-2, cleaves hemojuvelin decreasing the BMP-SMAD signaling axis, and thus inhibiting hepcidin expression [23]. Inhibition of the expression of this inhibitory protein may provide an attractive pathway for increasing hepcidin expression in beta-thalassemia patients [24], although some drawbacks exist [25]. A further hepcidin stimuli is inflammation, and this pathway is mainly modulated through the inflammatory cytokine IL-6 which activates the JAK-STAT3 pathway leading to increased hepcidin expression in the liver [26, 27].
To maintain iron homeostasis, the negative regulation of hepcidin expression is an important mechanism to ensure the availability of iron for biological activities. The erythropoietic cells of the bone marrow are the main consumers of iron provided by transferrin, and erythropoiesis is wholly dependent upon this source of iron. During erythropoiesis, erythroid cells secrete a factor or factors that suppress hepcidin expression in liver cells. This results in increased ferroportin activity and the transfer of iron from cellular stores to transferrin, thus supplying the demand for iron during erythropoiesis. The suppression of hepcidin also increases absorption of dietary iron [22]. The factor or factors secreted by erythroid cells remain to be clearly elucidated. Studies have implicated the growth and differentiation factor 15 (GDF15) which is known to be highly elevated in beta-thalassemia patients [28]. Other studies have suggested that the twisted gastrulation protein (TWSG1) may be the primary factor regulating hepcidin suppression in liver cells [29]. Moreover, erythroferrone has been recently identified and proposed as a candidate for suppressing hepcidin expression during erythropoiesis[30].
3. Thalassemia syndromes
Thalassemia syndromes are a group of inherited hematological disorders that constitute a major public health problems worldwide [31]. The term “thalassemia”, which has been used to described autosomal recessive anemic disorders, is derived from the Greek word “thalassa”(the Mediterranean sea) and “haima” (blood) since it was first applied to the anemias frequently encountered in people around the Mediterranean sea, particularly in Italy and the Greek coast and nearby islands [32]. Thalassemia syndromes are a heterogeneous group of anemias which are caused by genetic defects in globin genes. Defect of one or more globin genes cause a partial reduction or total depletion of globin chains synthesis thereby leading to inadequate production of hemoglobin [33]. The major types of thalassemia are alpha- and beta-thalassemia which are classified according to the nature of the defective globin [33].
4. Beta-thalassemia
Beta-thalassemia is a heterogeneous group of disorders leading to decreased or absent beta-globin production. A genetic defect of one or two beta-globin genes, which are located on chromosome 11 (p15.5), is the cause of beta-thalassemia [34]. To date, more than 200 point mutations have been identified in beta-globin genes and the immediate flanking regions [35]. The expression of mutated beta-globin genes can result in reduced or absent beta-globin production, unlike the large gene deletions in alpha-globin gene which solely result in loss of function [34]. According to these finding, beta-thalassemia can be phenotypically classified into 2 types; beta0-thalassemia where no beta-globin chains are synthesized and beta+-thalassemia where some beta-globin chains are synthesized [36]. In beta+-thalassemia, there is a 5% to 30 % reduction of beta-globin chains from normal levels [37]. The hallmark of beta-thalassemia is the reduced production or absence of Hb A (alpha2beta2), reactivation of Hb F (alpha2gamma2), and importantly, accumulation of excess alpha-globin chains which appears to underlie the main physiopathology of the disease. Patients with the most severe form of beta-thalassemia (beta0/beta0) develop serious microcytic anemia due to severe hemolysis and impaired production of new RBCs [38]. Bone deformity as a result of erythroid hyperplasia and enlargement of liver and spleen are also observed [35]. In heterozygous beta-thalassemia (beta/beta0,beta/beta+) or beta-thalassemia traits, Hb A formation is substantial due to the output from the remaining intact beta-globin gene, thus resulting in a nearly asymptomatic presentation with mild hypochromia with microcytosis but with relatively little evidence of anemia, hemolysis, or impaired erythropoiesis.
The most common subgroup of beta-thalassemia is beta-thalassemia/ Hb E which accounts for almost 50% of the patients with severe betathalassemia worldwide [38]. This compound heterozygote is very common in many regions where HbE is predominant. Hemoglobin E (Hb E) is the structural hemoglobin variant which is the most common in Southeast Asia including Thailand (10% –50% of the population) [39, 40]. The betaE-globin gene produces only small amounts of betaE-globin chains, which is similar to some mutations causing beta+-thalassemia and therefore Hb E trait resembles a very mild beta+-thalassemia trait while Hb E homozygotes exhibit more microcytosis but are still asymptomatic [41] The severity of betathalassemia/Hb E generally depends on the co-inheritance of alphaglobin hemoglobinopathies as well as the level of Hb F. Although patients with beta+-thalassemia/Hb E develop a mild anemia with only a few clinical abnormalities, an extraordinarily wide clinical spectrum, ranging from a moderate to a severe form of anemia resembling homozygous beta0-thalassemia are observed in beta0-thalassemia/Hb E patients
4.1. Molecular pathogenesis of beta-thalassemia
The main pathophysiology of beta-thalassemia is caused by the unbalanced production of alpha-globin and beta-globin chains where alpha-globin chains appear to be in excess [35]. Unlike beta-globin chains, alpha-globin chains are unable to form stable tetramers thus free excess alpha-globin chains tend to form insoluble aggregates which precipitate within the developing erythroid cell. This results in the induction of apoptosis in the developing erythroid precursor at the polychromatophilic normoblast stage in a process termed ineffective erythropoiesis [45]. In the small percentage of erythroid cells that progress to maturation, the accumulation of free alpha-globin chains efficiently generates ROS and oxidative stress, resulting in RBC membrane damage and subsequently increased hemolysis [46]. In normal RBCs hemoglobin is reversibly oxidized to methemoglobin, with cytochrome b5 reductase mediating the reduction back to hemoglobin. However, free globin chains (both alpha and beta) are susceptible to oxidation to hemichromes which can become irreversibly modified [47], allowing the hemichrome iron to generate reactive oxygen species [48]. A large part of the difference in pathology between alpha- and beta-thalassemia arises from the fact that the excess beta-chains present in alpha-thalassemia can form a soluble tetramer (hemoglobin H, HbH) while the excess alpha-globin chains present in betathalassemia cannot, resulting in the deposition of insoluble aggregates in the RBC membranes. In beta-thalassemia therefore, the combination of ineffective erythropoiesis of the developing erythroid precursor cells and increased hemolysis of the mature RBC are the main causes of anemia in these patients. The anemia in these patients leads to a feedback loop that results in increased expansion of erythroid progenitors and accelerated erythroid differentiation [49]. The markedly increased erythropoiesis in betathalassemia has been reported in many studies either by prediction, based on ferrokinetic studies [50], by differential counting from bone marrow aspirates or by in vitro observation of CD34 hematopoietic progenitor culture [52-55]. The markedly increased erythropoiesis in beta-thalassemia has been reported in many studies either by prediction, based on ferrokinetic studies [50], by differential counting from bone marrow aspirates [51], or by in vitro observation of CD34 hematopoietic progenitor culture [52-55]. The marked expansion of the erythroid mass is a well documented feature of intermediate and severe beta-thalassemia cases which results in the generation of more distressing features such as organ enlargement and particularly bone deformity and fragility [42, 56, 57]. Ultrastructural studies using electron microscopy have shown that the precipitated alpha-globin chains are in both the cytoplasm and the nucleus [58] and begin to be present predominately in polychromatic erythroblasts [59-61]. Moreover, abnormal erythroid nuclei showing a partial loss of nuclear membrane and presence of intranuclear aggregates of alpha-globin chains have also been observed in bone marrow erythroblasts of homozygous beta-thalassemia patients [59]. These findings led to investigations of intramedullary death which showed later that programmed cell death or apoptosis clearly occurred in erythroid precursors of beta-thalassemia major or as detected by DNA ladder formation [62] and the outer externalization of phosphatidylserine (PS) to the membrane leaflet [63]. Additionally, in vitro studies have also demonstrated that apoptosis primarily occurs at the polychromatophilic normoblast stage [53], the intermediate stage of erythroid precursor differentiation where the alpha-globin chain aggregates appear to present [59]. Previous studies have demonstrated that heat shock protein 70 (HSP 70) interacts directly with excess free alpha-globin chains and is sequestered in the cytoplasm [64]. This prevents HSP70 from performing its normal physiological role of protecting GATA-binding factor 1 (GATA1) from proteolytic cleavage resulting in premature degradation of GATA1 and maturation arrest and apoptosis of polychromatic normoblasts [65]. The marked degree of anemia, due to ineffective erythropoiesis, combined with a considerable tissue hypoxia promote erythropoietin (EPO) production, which has been shown in several studies to be increased in beta-thalassemia/Hb E patients as compared to normal controls [52, 66-68]. Increased levels of EPO are believed to be the main factor driving the expansion of the erythroid mass. Extensive erythropoiesis induces a large erythropoietic mass which can be found in the bone marrow, liver and spleen, as well as at extramedullary sites.
4.2. Iron overload in beta-thalassemia
Severe cases of beta-thalassemia require regular blood transfusion to reduce the chronic anemia. Multiple blood transfusions, increased hemolysis of red blood cells and increased gastrointestinal iron absorption (Figure 1) lead to iron overload [69], and cardiomyopathy as a consequence of iron overloading is the most common cause of death in transfusion-dependent thalassemia patients [70]. The human body loses only 1 mg -2 mg iron per day, while a unit of transfused red blood cells contains approximately 200 mg of iron [38]. A patient who receives 25 units of blood per year, accumulates 5 g of iron each year in the absence of any iron chelation therapy. Excess iron is extremely toxic to all cells of the body and can cause serious and irreversible organic damage, such as cirrhosis, diabetes, heart disease, and hypogonadism which lead to significant morbidity and mortality if untreated [69]. The iron overload on the body can be estimated by means of serum ferritin, hepatic iron concentration, urinary iron excretion and TIBC levels [71]. Threshold values for iron toxicityare a liver iron concentration exceeding 440 mmoles/g dry weight, serum ferritin >2 500 ng/mL, urinary iron excretion >20 mg/day, and transferrin saturation >75% [72]. The estimation of hepatic iron concentration by magnetic resonance imaging (MRI) is the most commonly employed test to evaluate iron overload in thalassemia major [73]. Increased iron overload has also been reported in patients with non-transfusion dependent thalassemia (NTDT)[74]. Beta-thalassemia carriers and patients who have a histidine to aspartic acid (H63D) mutation at codon 63 of the HFE gene show iron overloading, suggesting that the H63D mutation may have a modulating effect on iron absorption [75, 76].
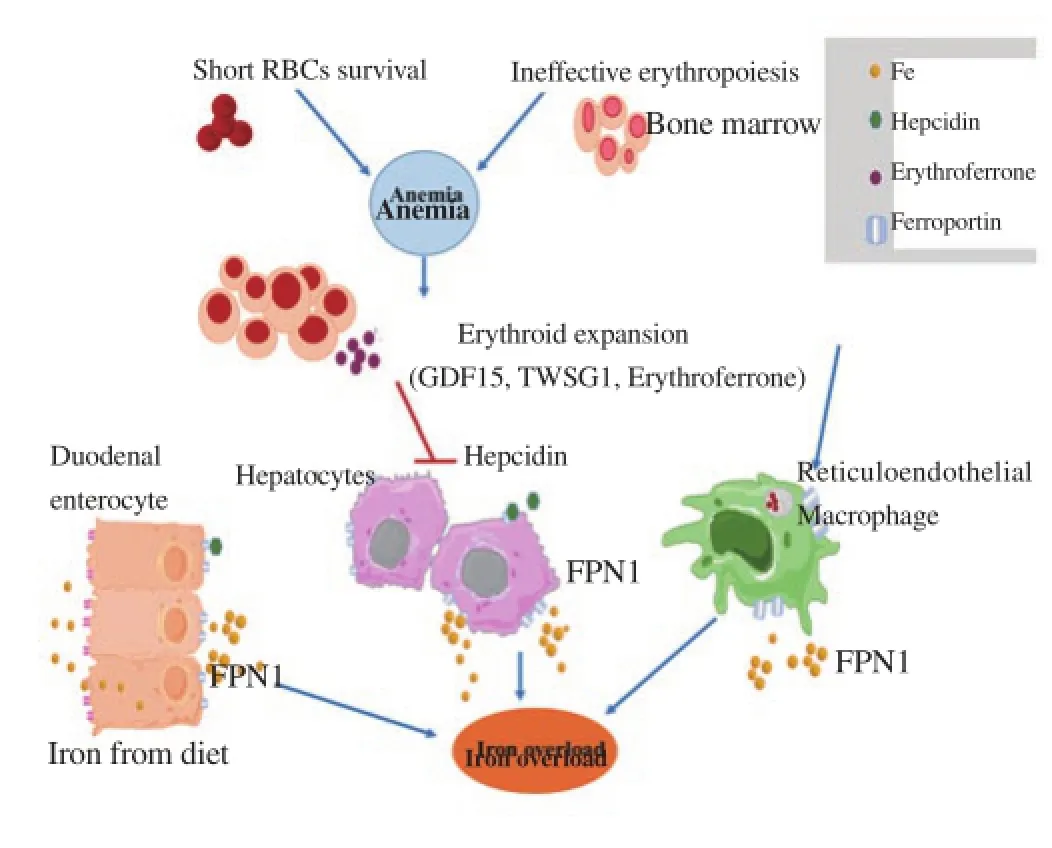
Figure 1. Proposed mechanism of iron dysregulation in beta-thalassemia disease.
Beta-thalassemia patients experience anemia mainly from ineffective erythropoiesis and shortened red blood cell (RBC) survival. The anemia induces erythropoietin (EPO) production leading to enhanced erythropoiesis. The dramatically increased erythroid expansion activates secretion of erythroid factors including growth differentiation factor 15 (GDF15), twisted-gastrulation 1 (TWSG1) and erythroferrone (ERFE). Excessive erythroid factors suppress hepcidin expression in hepatocytes resulting in increased iron absorption from duodenal enterocytes, the release of iron from the liver and the reticuloendothelial system, culminating in iron overload.
4.2.1. Pathophysiology of iron overload in beta-thalassemia
The excess iron in beta-thalassemia patients saturates the ability of the transferrin iron transport system, leading to non-transferrin bound iron (NTBI) and labile plasma iron (LPI) starting to circulate in plasma and subsequently becoming deposited inside susceptible cells [72]. Rather than using the transferrin receptor, NTBI enters cells by other cellular channels including L-type voltage-dependent Ca2+channel (LVDCC), a promiscuous divalent cation transporter [77] and Zip14, a member of the SLC39A zinc transporter family [78]. Long-term uptake and accumulation of NTBI and LIP, its redox active component, leads to increased levels of storage iron and labile cellular iron [79]. Tissues susceptible to iron accumulation by this mechanism include the liver, endocrine system and myocardium [80]. When the magnitude of the cellular labile iron pool exceeds the capacity of the cell to synthesize new ferritin molecules, a critical concentration is reached that can generate reactive oxygen species (ROS). ROS produced by the metabolism of NTBI plays a central role in inducing cellular dysfunction, apoptosis, and necrosis [81]. A variety of ROS, most notably hydroxyl radicals, increase lipid peroxidation and organelle damage, leading to cell death and fibrogenesis mediated by transforming growth factor beta1 (TGF-beta1) [82]. An underappreciated effect of iron overload is increased infection risk that is a high cause of mortality in beta-thalassemia patients [83] Oxidative stress is a major inducer of autophagy, which is important in the removal of oxidized proteins and damaged mitochondria. The increased activation of autophagy has been reported in beta-thalassemia/Hb E erythroblasts as compared to normal control erythroblasts, [84] suggesting that high levels of autophagy in beta-thalassemia/Hb E erythroblasts might be induced by ROS that contribute to the increased levels of apoptosis that lead to ineffective erythropoiesis in beta-thalassemia/Hb E erythroblasts [84].
Recently, dysregulation of ferroportin mRNA been reported in beta-thalassemia/ Hb E. While erythroblasts from normal controls show increased expression of ferroportin expression during differentiation under iron overload conditions, erythroblasts from beta-thalassemia/Hb E patients show no increase in ferroportin expression under the same growth conditions [85]. Thus the ability of these critical erythroid cells to export excess iron is curtailed, adding to the direct biological consequences of iron overload.
Iron overload can also contribute to ineffective erythropoiesis to a varying extent depending on the disorder (Figure 1). It has been suggested that the production of growth differentiation factor 15 (GDF15) [86, 87] and possibly other proteins, such as twistedgastrulation 1 (TWSG1) [29], contributes to the inhibition of hepcidin synthesis and thus promotes iron absorption in beta-thalassemia patients. Kautz and colleagues suggested that, upon increased erythropoiesis, bone marrow and spleen erythroblasts increasingly produce erythroferrone, which, upon secretion into the circulation, directly acts on the liver to inhibit hepcidin production and mediates increased iron mobilization and absorption during periods of erythropoietic stress [30].
4.3. Hepcidin expression in beta-thalassemia
Deficits in hepcidin expression in relation to thalassemia were first reported in a mouse model system (C57BI/6 Hbbth3/+) of severe anemia [88], and since then hepcidin levels in beta-thalassemia/Hb E disease,, beta-thalassemia trait and Hb E trait carriers have been reported [89]. Levels of hepcidin in beta-thalassemia patients have been shown to be extremely low [90-93] and serum from beta-thalassemia patients has been shown to suppress hepcidin expression in liver cells [86]. This will lead to continual, effectively unregulated absorption of dietary iron, leading to overloading. Liver hepcidin mRNA expression in patients with thalassemia major and thalassemia intermedia is inversely correlated with soluble transferrin receptor (sTfR) and erythropoietin (EPO), but not with iron stores [94]. The study proposed that hepcidin suppression in beta-thalassemia/Hb disease is associated with iron loading, saturation of iron binding proteins, and consequently, organ damage as indicated by an inverse association between hepcidin and NTBI across all patients, as well as correlation of NTBI and ferritin or LIC, and correlation of iron loading with ALT, an enzymatic marker of hepatic damage [89]. While moderate suppression of hepcidin with enhanced iron absorption was also observed in beta-thalassemia carriers; this was not the case with Hb E trait carriers [89,95]. The coinheritance of alpha-thalassemia results in a reduction in erythropoiesis and ameliorates hepcidin suppression [89]. Less severe forms of ineffective erythropoiesis, as observed in alphathalassemia, may cause late-onset and milder iron overload.
4.4. Hepcidin in the pathogenesis of beta-thalassemia
Anemia, tissue hypoxia and increased EPO production observed in beta-thalassemia promote the suppression of hepcidin and increase iron absorption in response to the demand for iron by erythroblasts [96]. Several hepcidin inhibitors released from erythroblasts during the process of differentiation have been proposed to regulate hepcidinexpression in beta-thalassemia. The cytokine members of the TGF-beta family, namely growth differentiation factor 15 (GDF15) was shown to be up-regulated in serum from thalassemia patients and can suppress hepcidin expression in hepatoma cells or in isolated human hepatocytes [28, 97]. Serum levels of this cytokine are strikingly elevated in patients with homozygous beta-thalassemia, while intermediate levels are found in carriers of alpha-thalassemia and in beta-thalassemia trait carriers. In contrast, sickle cell patients whose anemia is related to chronic haemolysis rather than ineffective erythropoiesis, show no or only modest GDF15 elevation [28]. TWSG1 is a second erythroid factor that has been identified as a hepcidin regulator. Levels of this protein are increased in the bone marrow, spleen and liver of thalassemic mice [29]. However, the level of TWSG1 in the serum of thalassemia patients remains to be reported. Hepcidin inhibition by the liver serine protease TMPRSS6 has also been shown [23, 98]. More recently, the hormone erythroferrone (ERFE) has been identified as a new erythroid regulator of hepcidin synthesis [30, 99]. In mouse models of intermediate thalassemia, bone marrow ERFE expression is increased in response to erythropoietin and mediates hepcidin suppression during stress erythropoiesis. ERFE-deficient mice fail to suppress hepcidin rapidly after hemorrhage and mice exhibit a delay in recovery from blood loss [99]. However, the molecular mechanisms underlying the suppression of hepcidin by these erythroid factors, as well as the interplay between the factors remain to be clarified.
4.5. Therapeutic targeting of hepcidin in beta-thalassemia
The standard treatment of severe beta-thalassemia is currently based on blood transfusions, iron chelation and splenectomy, allowing an increased survival and amelioration of the patients' quality of life [100]. A cure for inherited beta-thalassemia can be achieved by allogeneic hematopoietic stem cell transplantation, but the need to control transplant-related complications and the requirement for matched donors make this option available to only some patients [101], and as many as 60% of patients lack a suitable donor [101]. Alternative treatments, such as gene therapy or the induction of fetal hemoglobin (Hb F) are promising [102], but have yet to make it to the bedside. As iron overload is the most important complication for the patients with blood transfusion, iron chelation is essential to control iron overload and its toxicity [103] and effective management of iron overload in thalassemia requires monitoring both for iron toxicity and the effects of excessive chelation. Recently however, improved knowledge of the relationships between iron overload and hepcidin has led to the development of novel approaches that target the pathophysiology of the disease with the aim of reducing iron overload and, at the same time, of alleviating ineffective erythropoiesis. Hepcidin levels are low in thalassemia patients with concomitant pathophysiological consequences, and the restoration of hepcidin to normal levels is an attractive novel therapeutic strategy. Studies in a mouse model of beta-thalassemia have shown that increasing hepcidin reduces iron bioavailable to erythroblasts, resulting in decreased heme synthesis and improved erythroid precursor and reticulocyte survival [104]. Similarly small synthetic peptides (minihepcidins) can decrease serum iron, prevent iron overload and promote iron redistribution in hepcidin-deficient mice [105]. In another approach, administration of BMP6, the natural ligand of the BMP receptor involved in hepcidin regulation, was shown to activate the hepcidin transcription regulated pathway and to correct the high iron saturation and iron maldistribution in a HFE model of hereditary hemochromatosis [106]. Transgenic inactivation of the membrane protease TMPRSS6 in HFE mice increased hepcidin expression and reversed their iron overload phenotype, suggesting that the administration of a specific inhibitor of the enzymatic activity of TMPRSS6 could be used to treat iron overload [107, 108]. An RNAi therapeutic targeting Tmprss6 has been shown to decrease iron overload with diminished hepcidin expression and may have efficacy in modifying disease-associated morbidities of thalassemia [24]. A combination therapy of RNAi against Tmprss6 together with administration of the iron chelater deferiprone has been reported to result in a significant reduction of liver iron content and improved erythropoietic efficiency in thalassemic mice [109, 110].
5. Conclusions
While the genetic lesions engendering beta-thalassemia are well characterized, how these lesions lead to the full pathological spectrum of the disease remain less well understood. While deposition of unpaired alpha-globin chains during erythropoiesis is a major event, it is clear that dysregulation of iron homeostasis in both independant and transfusion dependent and transfusion independent beta-thalassemia patients is a dominant physiological effect. Understanding hepcidin expression and regulation in the context of the beta-thalassemia patient is vital to developing rational therapeutic interventions to provide safe, effective and lifelong treatments.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Acknowledgments
DRS is supported by the Thailand Research Fund (BRG5780004, IRG5780009 and IRN58W0002) and by a Research Chair Grant from the National Science and Technology Development Agency (NSTDA). KL is supported by the Thailand Research Fund (MRG5980160). WS is supported by a Thai Royal Golden Jubilee Ph. D. Research Scholarship (PHD/0101/2553). The funders had no role in the design, analysis and interpretation of this article or in the decision to publish.
References
[1] MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Sign 2008; 10(6): 997-1030.
[2] Kaplan J, Ward DM, De Domenico I. The molecular basis of iron overload disorders and iron-linked anemias. Int J Hematol 2011; 93(1): 14-20.
[3] Fuqua BK, Vulpe CD, Anderson GJ. Intestinal iron absorption. J Trace Elem Med Biol 2012; 26(26): 115-119.
[4] Hershko C, Graham G, Bates GW, Rachmilewitz EA. Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br J Haematol 1978; 40(2): 255-263.
[5] Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta 2012; 1823(1823): 1434-1443.
[6] Conrad ME, Umbreit JN, Moore EG, Hainsworth LN, Porubcin M, Simovich MJ, Nakada MT, Dolan K, Garrick MD. Separate pathways for cellular uptake of ferric and ferrous iron. Am J Physiol Gastrointest Liver Physiol 2000; 279(4): 767-774.
[7] Fleming MD, Trenor CC, 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 1997; 16(4): 383-386.
[8] San Martin CD, Garri C, Pizarro F, Walter T, Theil EC, Nunez MT. Caco-2 intestinal epithelial cells absorb soybean ferritin by mu2 (AP2)-dependent endocytosis. J Nutr 2008; 138(4): 659-666.
[9] Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000; 275(26): 19906-19912.
[10] Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000; 403(6771): 776-781.
[11] McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000; 5(2): 299-309.
[12] Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, et al. Identification of a ferrireductase required for efficient transferrindependent iron uptake in erythroid cells. Nat Genet 2005; 37(11): 1264-1269.
[13] Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood 2006; 108(4): 1388-1394.
[14] Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 2003; 102(12): 4191-4197.
[15] Liu XB, Hill P, Haile DJ. Role of the ferroportin iron-responsive element in iron and nitric oxide dependent gene regulation. Blood Cells Mol Dis 2002; 29(3): 315-326.
[16] Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/ IREG1/MTP1 gene expression. J Hepatol 2003; 39(5): 710-715.
[17] Cianetti L, Segnalini P, Calzolari A, Morsilli O, Felicetti F, Ramoni C, Gabbianelli M, Testa U, Sposi NM. Expression of alternative transcripts of ferroportin-1 during human erythroid differentiation. Haematologica 2005; 90(12): 1595-1606.
[18] Zhang D-L, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metabolism 2009; 9(5): 461-473.
[19] Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, et al. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J Biol Chem 2009; 284(36): 24155-24167.
[20] Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276(276): 7806-7810.
[21] Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol 2009; 122(2-3): 78-86.
[22] Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schumann K, Debnam ES, Srai SK. Effect of hepcidin on intestinal iron absorption in mice. Blood 2004; 103(10): 3940-3944.
[23] Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab 2008; 8(6): 502-511.
[24] Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, Hettinger J, Bumcrot D, Fleming MD. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013; 121(7): 1200-1208.
[25] Silvestri L. Inhibiting the hepcidin inhibitor for treatment of iron overload. Blood 2013; 121(7): 1068-1069.
[26] Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113(9): 1271-1276.
[27] Sakamori R, Takehara T, Tatsumi T, Shigekawa M, Hikita H, Hiramatsu N, Kanto T, Hayashi N. STAT3 signaling within hepatocytes is required for anemia of inflammation in vivo. J Gastroenterol 2010; 45(2): 244-248.
[28] Tanno T, Bhanu NV, Oneal PA, Goh S-H, Staker P, Lee YT, Moroney JW, Reed CH, Luban NLC, Wang R-H, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007; 13(9): 1096-1101.
[29] Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, Bhupatiraju A, Lee YT, Goodnough JB, Harandi O, Ganz T, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009; 114(1): 181-186.
[30] Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014; 46(7): 678-684.
[31] Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet 2012; 379(9813): 373-383.
[32] Marks PA, Bank A. Molecular pathology of thalassemia syndromes. Fed Proc 1971; 30(30):977-982.
[33] Villegas A, Ropero P, Gonzalez FA, Anguita E, Espinos D. The thalassemia syndromes: molecular characterization in the Spanish population. Hemoglobin 2001; 25(3): 273-283.
[34] Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med.2005; 353(11): 1135-1146.
[35] Olivieri NF. The beta-thalassemias. N Engl J Med 1999; 341(2): 99-109.
[36] Ho PJ, Thein SL. Gene regulation and deregulation: a beta globin perspective. Blood Rev 2000; 14(2): 78-93.
[37] R.Gibbons DRH, Nancy F. Olivieri, Wood WG. Thalassaemia syndromes. In: D.J.Weatherall JBC. Theβ-thalassaemias. 4th ed. Oxford: Blackwell Science; 2001,p. 287-356.
[38] Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the beta-Thalassemias. Cold Spring Harb Perspect Med 2012; 2(12): a011726-a011726.
[39] Fucharoen. S. Disorders of hemoglobin: genetics, pathophysiology andclinical management. 3 ed. New York: Cambridge University Press; 2001, p. 1147-1148.
[40] Fucharoen S, Winichagoon P, Pootrakul P, Piankijagum A, Wasi P. Variable severity of Southeast Asian beta 0-thalassemia/Hb E disease. Birth Defects Orig Artic Ser 1987; 23(5): 241-248.
[41] Fairbanks VF, Oliveros R, Brandabur JH, Willis RR, Fiester RF. Homozygous hemoglobin E mimics beta-thalassemia minor without anemia or hemolysis: hematologic, functional, and biosynthetic studies of first North American cases. Am J Hematol 1980; 8(1): 109-121.
[42] Fucharoen S. Disorders of hemoglobin: Genetics, pathophysiology, and clinical management. In: Steinberg MH FB, Higgs DR, Nagel RL. (eds.) Hemoglobin E disorders. Cambridge: Cambridge University Press; 2001, p.1139-1154.
[43] Sripichai O, Whitacre J, Munkongdee T, Kumkhaek C, Makarasara W, Winichagoon P, Abel K, Braun A, Fucharoen S. Genetic analysis of candidate modifier polymorphisms in Hb E-beta 0-thalassemia patients. Ann N Y Acad Sci 2005; 1054(1): 433-438.
[44] Winichagoon P, Thonglairoam V, Fucharoen S, Wilairat P, Fukumaki Y, Wasi P. Severity differences in beta-thalassaemia/haemoglobin E syndromes: implication of genetic factors. Br J Haematol 1993; 83(4): 633-639.
[45] Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, et al. Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp hematol 2000; 28(12): 1343-1353.
[46] Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol 2002; 9: 123-126.
[47] Rachmilewitz EA, Peisach J, Bradley TB, Blumberg WE. Role of haemichromes in the formation of inclusion bodies in haemoglobin H disease. Nature 1969; 222(5190): 248-250.
[48] Rachmilewitz EA, Peisach J, Blumberg WE. Studies on the stability of oxyhemoglobin A and its constituent chains and their derivatives. J Biol Chem 1971; 246(10): 3356-3366.
[49] Arlet JB, Dussiot M, Moura IC, Hermine O, Courtois G. Novel players in beta-thalassemia dyserythropoiesis and new therapeutic strategies. Curr Opin Hematol 2016; 23(3): 181-188.
[50] Pootrakul P, Sirankapracha P, Hemsorach S, Moungsub W, Kumbunlue R, Piangitjagum A, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood 2000; 96(7): 2606-2612.
[51] Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood 2000; 96(10): 3624-3629.
[52] Chen JS, Lin KH, Tsao CJ. Peripheral blood hematopoietic progenitor cells in beta-thalassemia major. Int J Cell Cloning 1992; 10(6): 338-343.
[53] Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, et al. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol 2000; 28(12): 1343-1353.
[54] Kittikalayawong Y, Sila-asna M, Bunyaratvej A. Enhanced maturation and proliferation of beta-thalassemia/Hb E erythroid precursor cells in culture. Southeast Asian J Trop Med Public Health 2005; 36(5): 1298-1303.
[55] Mai A, Jelicic K, Rotili D, Di Noia A, Alfani E, Valente S, et al. Identification of two new synthetic histone deacetylase inhibitors that modulate globin gene expression in erythroid cells from healthy donors and patients with thalassemia. Mol Pharmacol 2007; 72(5): 1111-1123.
[56] Weatherall DJ JBC. Thalassaemia syndromes. In: Weatherall DJ JBC. (eds.) The pathophysiology of the thalassaemias. 4th ed. Oxford: Blackwell Science; 2001, p. 192-236.
[57] Nancy F. Olivieri DJW. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. In: Steinberg MH FB, Higgs DR, Nagel RL. (eds.) Clinical aspects of β-Thalassemia. Cambridge: Cambridge University Press; 2001, p. 277-341.
[58] Polliack A, Yataganas X, Rachmilewitz EA. Ultrastructure of the inclusion bodies and nuclear abnormalities in beta-thalassemic erythroblasts. Ann N Y Acad Sci 1974; 232(6869): 261-282.
[59] Wickramasinghe SN, Bush V. Observations on the ultrastructure of erythropoietic cells and reticulum cells in the bone marrow of patients with homozygous beta-thalassaemia. Br J Haematol 1975; 30(4): 395-399.
[60] Wickramasinghe SN, Hughes M, Wasi P, Fucharoen S, Modell B. Ineffective erythropoiesis in haemoglobin E beta -thalassaemia: an electron microscope study. Br J Haematol 1981; 48: 451-457.
[61] Modell CB. Haemoglobinopathies. The pathophysiology of betathalassaemia major. J Clin Pathol Suppl (R Coll Pathol) 1974; 8(suppl 8): 12-18.
[62] Yuan J, Angelucci E, Lucarelli G, Aljurf M, Snyder LM, Kiefer CR, et al. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley's anemia). Blood 1993; 82(2): 374-377.
[63] Borenstain-Ben Yashar V, Barenholz Y, Hy-Am E, Rachmilewitz EA, Eldor A. Phosphatidylserine in the outer leaflet of red blood cells from beta-thalassemia patients may explain the chronic hypercoagulable state and thrombotic episodes. Am J Hematol 1993; 44(1): 63-65.
[64] Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, et al. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature 2002; 417(6890): 758-763.
[65] Arlet JB, Ribeil JA, Guillem F, Negre O, Hazoume A, Marcion G, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in beta-thalassaemia. Nature 2014; 514(7521): 242-246.
[66] Chaisiripoomkere W, Jootar S, Chanjarunee S, Ungkanont A. Serum erythropoietin levels in thalassemia major and intermedia. Southeast Asian J Trop Med Public Health 1999; 30(4): 786-788.
[67] Nisli G, Kavakli K, Aydinok Y, Oztop S, Cetingul N. Serum erythropoietin levels in patients with beta thalassemia major and intermedia. Pediatr Hematol Oncol 1997; 14(2): 161-167.
[68] Paritpokee N, Wiwanitkit V, Bhokaisawan N, Boonchalermvichian C, Preechakas P. Serum erythropoietin levels in pediatric patients with betathalassemia/hemoglobin E. Clin Lab 2002; 48(11-12): 631-634.
[69] Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011; 118(16): 4321-4330.
[70] Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Survival and complications in thalassemia. Ann N Y Acad Sci 2005; 1054(1): 40-47.
[71] Mishra AK, Tiwari A. Iron overload in Beta thalassaemia major and intermedia patients. Maedica (Buchar) 2013; 8(8): 328-332.
[72] Hershko C. Pathogenesis and management of iron toxicity in thalassemia. Ann N Y Acad Sci 2010; 1202: 1-9.
[73] Henninger B, Kremser C, Rauch S, Eder R, Zoller H, Finkenstedt A, et al. Evaluation of MR imaging with T1 and T2* mapping for the determination of hepatic iron overload. Eur Radiol 2012; 22(11): 2478-2486.
[74] Musallam KM, Cappellini MD, Taher AT. Iron overload in betathalassemia intermedia: an emerging concern. Curr Opin Hematol 2013; 20(3): 187-192.
[75] Melis MA, Cau M, Deidda F, Barella S, Cao A, Galanello R. H63Dmutation in the HFE gene increases iron overload in beta-thalassemia carriers. Haematologica 2002, 87(3): 242-245.
[76] Longo F, Zecchina G, Sbaiz L, Fischer R, Piga A, Camaschella C. The influence of hemochromatosis mutations on iron overload of thalassemia major. Haematologica 1999; 84(9): 799-803.
[77] Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, et al. L-type Ca2+channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 2003; 9(9): 1187-1194.
[78] Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci 2006; 103(37): 13612-13617.
[79] Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol 2014; 5: 45.
[80] Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail 2010; 16(11): 888-900.
[81] Bresgen N, Eckl PM. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015; 5(2): 808-847.
[82] Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem 2013; 288(2): 770-777.
[83] Telfer PT, Warburton F, Christou S, Hadjigavriel M, Sitarou M, Kolnagou A, et al. Improved survival in thalassemia major patients on switching from desferrioxamine to combined chelation therapy with desferrioxamine and deferiprone. Haematologica 2009; 94(12): 1777-1778.
[84] Lithanatudom P, Wannatung T, Leecharoenkiat A, Svasti S, Fucharoen S, Smith DR. Enhanced activation of autophagy in beta-thalassemia/Hb E erythroblasts during erythropoiesis. Ann Hematol 2011; 90(7): 747-758.
[85] Sornjai W, Jaratsittisin J, Khungwanmaythawee K, Svasti S, Fucharoen S, Lithanatudom P, et al. Dysregulation of ferroportin gene expression in beta(0)-thalassemia/Hb E disease. Ann Hematol 2016; 95(3): 387-396.
[86] Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007; 13(9): 1096-1101.
[87] Tanno T, Noel P, Miller JL. Growth differentiation factor 15 in erythroid health and disease. Curr Opin Hematol 2010; 17(3): 184-190.
[88] Adamsky K, Weizer O, Amariglio N, Breda L, Harmelin A, Rivella S, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol 2004; 124(1): 123-124.
[89] Jones E, Pasricha SR, Allen A, Evans P, Fisher CA, Wray K, et al. Hepcidin is suppressed by erythropoiesis in hemoglobin E betathalassemia and beta-thalassemia trait. Blood 2015; 125(5): 873-880.
[90] Gardenghi S, Grady RW, Rivella S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol Oncol Clin North Am 2010; 24(6): 1089-1107.
[91] Gardenghi S, Ramos P, Follenzi A, Rao N, Rachmilewitz EA, Giardina PJ, Grady RW, Rivella S. Hepcidin and Hfe in iron overload in betathalassemia. Ann N Y Acad Sci 2010; 1202(1): 221-225.
[92] Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with betathalassemia major: a longitudinal study. Blood 2013; 122(1): 124-133.
[93] Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, Goldberg YP, Sakellaropoulos N, Ganz T, Nemeth E. Hepcidin in iron overload disorders. Blood 2005; 105(10): 4103-4105.
[94] Kattamis A, Papassotiriou I, Palaiologou D, Apostolakou F, Galani A, Ladis V, et al. The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica 2006; 91(6):809-812.
[95] Guimaraes JS, Cominal JG, Silva-Pinto AC, Olbina G, Ginzburg YZ, Nandi V, et al. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol 2015; 94(6): 511-518.
[96] Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006; 108(12): 3730-3735.
[97] Weizer-Stern O, Adamsky K, Amariglio N, Levin C, Koren A, Breuer W, et al. Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Br J Haematol 2006; 135(1): 129-138.
[98] Folgueras AR, de Lara FM, Pendas AM, Garabaya C, Rodriguez F, Astudillo A, Bernal T, Cabanillas R, Lopez-Otin C, Velasco G. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008; 112(6): 2539-2545.
[99] Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, Nemeth E, Ganz T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015; 126(17): 2031-2037.
[100] Taher AT, Viprakasit V, Musallam KM, Cappellini MD. Treating iron overload in patients with non-transfusion-dependent thalassemia. Am J Hematol 2013; 88(5): 409-415.
[101] Lucarelli G, Isgro A, Sodani P, Gaziev J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med 2012; 2(5): 136-147.
[102] Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood 2013; 121(12): 2199-2212.
[103] Saliba AN, Harb AR, Taher AT. Iron chelation therapy in transfusiondependent thalassemia patients: current strategies and future directions. J Blood Med 2015; 6:197-209.
[104] Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest 2010; 120(12): 4466-4477.
[105] Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012; 120(18): 3829-3836.
[106] Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010; 139(5): 1721-1729.
[107] Nai A, Pagani A, Silvestri L, Campostrini N, Corbella M, Girelli D, et al. TMPRSS6 rs855791 modulates hepcidin transcription in vitro and serum hepcidin levels in normal individuals. Blood 2011; 118(16): 4459-4462.
[108] Finberg KE, Whittlesey RL, Andrews NC. Tmprss6 is a genetic modifier of the Hfe-hemochromatosis phenotype in mice. Blood 2011; 117(17): 4590-4599.
[109] Casu C, Aghajan M, Oikonomidou PR, Guo S, Monia BP, Rivella S. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica 2014; 124(1): e8-e11.
[110] Schmidt PJ, Racie T, Westerman M, Fitzgerald K, Butler JS, Fleming MD. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am J Hematol 2015; 90(4): 310-313.
Document heading 10.1016/j.apjtm.2016.07.035
23 June 2016
in revised form 24 July 2016
Dr Kamonlak Leecharoenkiat, Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, 154 Rama 4 Road, Bangkok, Thailand 10330
Tel: +662-2181086 ext. 334.
Fax: +662-2181064
✉Molecular Pathology Laboratory, Institute of Molecular Biosciences, Mahidol University, 25/25 Phuttamonthon 4 Road, Salaya, Nakhon Pathom 73170, Thailand.
E-mail: duncan_r_smith@hotmail.com
Tel: 66 (0) 2441-9003 to 7.
Fax: 66 (0) 2441-1013.
Foundation project: DRS is supported by the Thailand Research Fund (BRG5780004, IRG5780009 and IRN58W0002) and by a Research Chair Grant from the National Science and Technology Development Agency (NSTDA). WS is supported by a Thai Royal Golden Jubilee Ph. D. Research Scholarship (PHD/0101/2553). The founders had no role in the design, analysis and interpretation of this article or in the decision to publish.
 Asian Pacific Journal of Tropical Medicine2016年11期
Asian Pacific Journal of Tropical Medicine2016年11期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Modifiable determinants of attitude towards dengue vaccination among healthy inhabitants of Aceh, Indonesia: Findings from a communitybased survey
- Clinical significance of dynamic detection for serum levels of MCP-1, TNF-α and IL-8 in patients with acute pancreatitis
- Expression and mechanism of action of miR-196a in epithelial ovarian cancer
- Protective effect of antioxidant on renal damage caused by Doxorubicin chemotherapy in mice with hepatic cancer
- Mechanism of action of Zhuyu Annao pill in mice with cerebral intrahemorrhage based on TLR4
- Acetylcholinesterase, butyrylcholinesterase and paraoxonase 1 activities in rats treated with cannabis, tramadol or both