
基于密度泛函理论的CuO氧载体释氧机理研究*
2016-11-10 08:13冯晓鸣冯永新刘亚明李方勇
新能源进展 2016年5期
冯晓鸣,冯永新,刘亚明,李方勇
(广东电网有限责任公司电力科学研究院,广州 510080)
基于密度泛函理论的CuO氧载体释氧机理研究*
冯晓鸣,冯永新,刘亚明†,李方勇
(广东电网有限责任公司电力科学研究院,广州 510080)
化学链氧解耦(CLOU)是基于化学链燃烧(CLC)技术的一种新型燃烧方式,具有CO2内分离的优良特性。具有良好吸氧释氧性能的氧载体是CLOU技术的关键,其宏观层面的反应性决定于其微观层面的晶格氧传输机理,但目前对这种微观机理的研究非常缺乏。本文采用量子化学计算方法——密度泛函理论(DFT)研究 CuO氧载体释氧的微观机理,构建了CuO团簇及CuO平板模型,模拟团簇及表面的释氧过程。CuO平板模型释氧包括O原子在CuO内部扩散、表面O2的形成及释放过程。结果表明:CuO(111)表面释氧过程的最高能量势垒为3.16 eV,与实验值3.39 eV接近,低于CuO团簇模型的释氧能量势垒3.51 eV;此外,O原子在CuO(111)内部的扩散势垒仅为0.87 eV,说明CuO(111) 释氧的限制步骤是表面O2的形成过程。
密度泛函理论(DFT);化学链氧解耦;铜基氧载体;释氧动力学
0 前 言
化学链氧解耦(chemical looping with oxygen uncoupling,CLOU)是基于化学链燃烧(chemical looping combustion,CLC)提出的一种新型的燃烧技术。传统的CLC中燃料的燃烧借助于氧载体传递的活性晶格氧,而CLOU中燃料的燃烧借助于氧载体释放的O2分子,可以大大加快燃料与氧载体的反应速率。CLOU过程可分为三步:首先是氧载体MeOx在燃料反应器中释放 O2(高温);随后燃料与氧载体释放的O2发生反应,生成CO2和水蒸气,水蒸气被冷凝之后便可得到高纯度的 CO2,同时氧载体逐渐转变为低氧势的 MeOx-1;最后,低氧势氧载体MeOx-1进入空气反应器中与空气发生反应,恢复为活性MeOx。燃料反应器与空气反应器释放的热量与燃料直接燃烧释放的热量相等。CLOU实现了CO2的内分离,降低了CO2捕集成本,并可避免快速型和热力型NOx的生成,在燃煤电站CO2低能耗捕集方面具有很好的应用前景。
实现 CLOU过程的关键是选择具有吸氧/释氧特性的氧载体,目前应用较多并已被证明的氧载体主要有金属Cu、Mn、Co的氧化物及钙钛矿型氧化物[1]。其中 Cu基氧载体(CuO/Cu2O)因为拥有较宽泛的释氧-吸氧温度区间(800℃~1 100℃)、优良的释氧-吸氧速率、合适的材料属性、较为廉价和环境友好等特点而受到广泛的关注。MATTISSON等[1-2]利用流化床反应器测试了冷冻成粒法制备的CuO/Al2O3和CuO/ZrO2氧载体的释氧性能及与CH4和石油焦的反应性能,证明了Cu基氧载体可快速分解并释放 O2,促进了 CH4和石油焦的燃烧。ADÁNEZ-RUBIO等[3]在串行流化床中用Cu基氧载体与不同煤阶的煤进行CLOU实验,尾气中没有检测到未燃尽气体,说明铜基氧载体CLOU过程可保证燃料完全燃烧。
目前CLOU中氧载体的释氧动力学研究多通过实验进行,例如通过流化床反应器或热重分析实验(TGA)获得宏观数据,对氧载体的释氧/吸氧性能进行总结分析,计算氧载体的活化能、指前因子等动力学数据,以此判定其反应性能;也可通过XRD、SEM等仪器检测氧载体的物相和微观形貌的变化,但这些实验方法仍很难对氧载体表面 O2分子的形成、氧载体内部原子的迁移等微观现象进行描述。而基于密度泛函理论(DFT)的量子化学计算方法从体系的微观结构、物理性质与宏观性能的因果关系出发,可对已有实验结果进行解释,进而设计性能良好的材料。例如开展对CLC中氧载体的氧传递特性的研究,量子化学计算方法可很好地辅助、指导实验进行。SIRIWARDANE等[4]基于DFT研究了C和CuO的作用机理:C接近CuO使Cu—O键伸长,逐渐造成Cu—O键断裂,活化了CuO中的O原子,促进了低温固-固反应的进行,很好地解释了TGA实验中低温下C与CuO反应生成CO2的过程。DONG等[5]基于DFT研究了CO与Fe2O3的反应机理,指出还原反应分为三步:Fe3++CO→Fe2++CO2、Fe2++CO→Fe++CO2、Fe++CO→Fe+CO2,得出反应物结构变化、电荷转移、能量势垒等,发现第二步反应的能量势垒远远大于第一步,说明为提高CLC中燃料反应速率,应避免 Fe2O3的过度还原。TAN等[6]研究了氧载体中Fe2O3与ZrO2的协同作用机理,DFT计算表明,ZrO2的加入可有效降低Fe2O3与CO反应的能量势垒,说明Fe2O3与ZrO2有良好的协同作用。LI等[7]结合TGA、SEM实验,用DFT方法解释了TiO2可以提高活性氧载体性能的原因,分别计算了三种氧载体FeO、FeTiO3和缺陷FeO中O2-的扩散势垒,发现添加TiO2可有效减小O2-的扩散势垒,与实验吻合,且O2-的扩散是反应速率的一个决定因素,影响氧载体的反应性能。已有文献多集中于化学链燃烧中氧载体与燃料的反应性能研究,对 CLOU中的氧载体的释氧吸氧性能研究鲜有报道。氧载体吸氧速率通常大于释氧速率,本文主要针对关键的释氧动力学进行研究,采用基于DFT的量子化学计算方法,系统研究CuO(111)表面和团簇中 O原子的扩散、O2分子形成及释放过程,探讨CuO释氧过程的限制步骤,并将计算活化能与实验值进行比较。
1 计算方法和模型
CuO为单斜晶体,属于C2/c空间群,晶格参数为a=4.690、b=3.420、c=5.131、β=99.54,其原胞结构如图1a。本文采用基于DFT的CASTEP程序包模拟了Cu4O4团簇模型和CuO(111)表面模型的释氧过程,其初始的优化结构分别如图1b、图1c所示。CuO团簇模型选取Cu4O4立方结构,CuO(111)表面选取3层(2×2)周期性平板结构(表面2层开放优化,底 1层固定),模型表面的真空层厚度设为20 Å,以避免不同层晶间的干扰。本文之所以选择CuO(111)表面为研究对象,是因为相比于其他低能量表面,CuO(111)表面能最小,说明其结构更稳定。此外,选择两种CuO模型是为了更好地模拟复杂的CuO氧载体结构。结构优化及能量计算过程中,体系交换相关能部分采用广义梯度近似(generalized gradient approximation,GGA)的 Perdew-Wang(PW91)方法[8]进行处理;体系布里渊区积分采用Monkhorst- Pack形式的特殊K点方法[9],K点网格数取(4×2×1)。结构优化以能量、位移和力收敛为判据,收敛阀分别为1×10-5eV、0.001 Å和0.03 eV/Å。截断能取400 eV,平衡计算代价与计算精度。在计算释氧过程的活化能时,采用程序中的 TS Search模块,并选取Complete LST/QST 方法对其进行过渡态搜索。反应过程的活化能可用下式计算:

其中 E(TS)为反应过渡态总能量,E(IS)为反应初始态总能量。
为了验证参数选择的合理性,并为之后的计算结果提供参考,本文首先计算了CuO原胞、Cu4O4团簇及自由O2分子优化后的结构信息,将计算值与实验值进行了比较,结果如表 1。可看出计算值与实验值符合非常好,误差均小于1%。
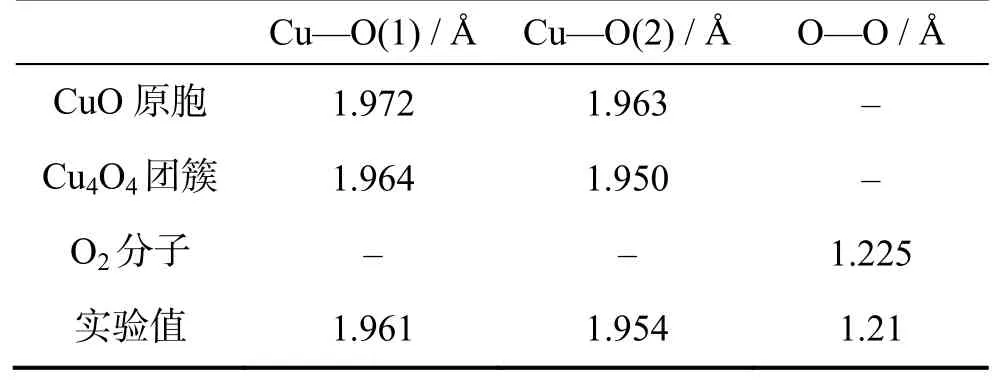
表1 CuO原胞、Cu4O4团簇及自由O2分子键长Table 1 The bond length of CuO unit cell,Cu4O4cluster and O2
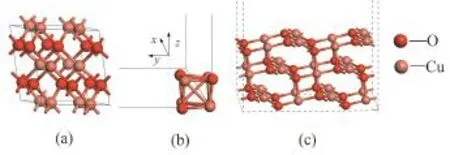
图1 计算模型:(a)CuO原胞模型;(b)Cu4O4团簇模型;(c)CuO(111)表面模型Fig. 1 Computing models: (a) CuO unit cell,(b) Cu4O4cluster,and (c) CuO(111) slab with different types of Cu and O atoms
2 结果与分析
2.1 团簇模型Cu4O4→Cu4O2+O2过程
反应初始态为优化后的立方团簇模型Cu4O4,用IS表示(如图2-IS),追踪的O原子用蓝色标记。以Cu4O2团簇及自由O2分子为反应产物模型(使两者间距离大约为 6 Å),对其进行结构优化得到 FS(如图2-FS)。在IS的基础上,移动上层两个O原子使之距离靠近,对结构进行优化得到了稳定的MS结构(如图2-MS),两个O原子成键并向上移动远离Cu4O2,成为整个反应的中间态。对IS→MS进行过渡态搜索得到过渡态TS1。IS→MS中,追踪的两个O原子与下层Cu原子的键首先断裂,之后两个O原子逐渐靠近,最终形成O—O键结构,其键长为1.427 Å,比单独优化自由O2分子的键长1.225 Å略长。说明这一反应阶段形成了 O2分子的初始结构,但仍吸附于Cu4O2上,需克服Cu—O键能,才能最终形成自由O2分子。对MS→FS进行过渡态搜索,得到过渡态TS2。这一反应阶段为O2分子脱附过程,首先上层Cu—O键断裂,O—O与Cu4O2团簇分离,O—O键长逐渐变短,最终缩短为FS中的1.259 Å,非常接近自由O2分子键长,说明O2分子已完全脱离Cu4O2。因此,整个Cu4O4→2Cu2O+O2过程包括O—O键形成阶段和O2分子脱附过程。
图 2同时给出了反应各个阶段的能量变化曲线,O—O键形成阶段的反应热为-1.15 eV,说明这一过程是放热反应,需克服的势垒为 0.84 eV。O2分子脱附阶段的反应热为3.50 eV,需克服的能量势垒为3.51 eV。这说明O2分子脱附需要吸收大量的热,可以通过O2与燃料反应放热来提供。从能量变化图可看出,O2分子脱附能量势垒远远大于O—O键形成势垒,说明团簇释氧过程的限制步骤为 O2脱附过程。因此,可通过某些途径降低O2分子脱附的能量势垒,来提高团簇CuO的释氧性能,其中一个可行的办法是添加其他负载物,如Al2O3、ZrO2,使负载与CuO团簇产生协同作用,进而减小Cu—O键断裂需克服的能量势垒。
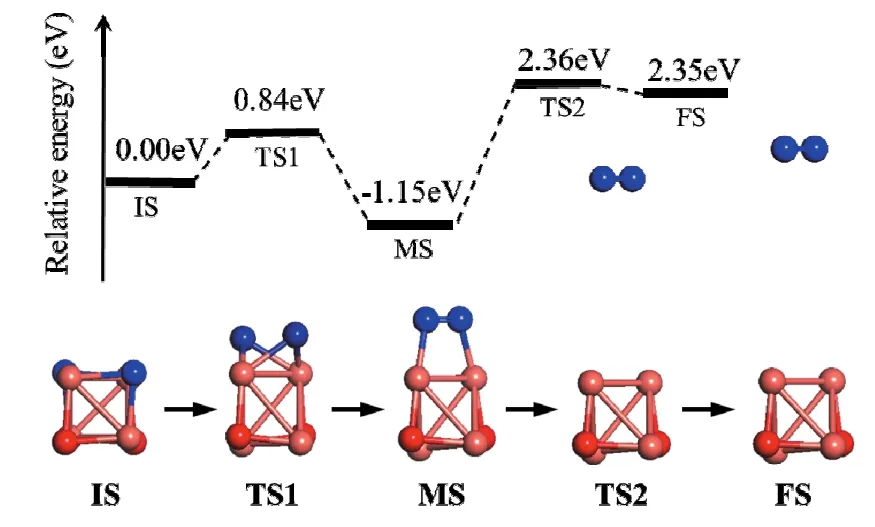
图2 Cu4O4团簇释氧过程的结构及能量变化Fig. 2 The structure and energy in Cu4O4oxygen release process
2.2 O2分子在CuO(111)表面的脱附过程
实际的氧载体颗粒通常比较复杂,但在微观上其释氧过程通常包括以下步骤:(1)氧载体表面的O原子在高温低氧分压的气氛中被活化;(2)活化的O原子结合,逐渐形成O2分子结构,并脱离CuO表面;(3)CuO内部氧负离子扩散至表面,提供O2分子形成需要的活化氧。本文以周期性 CuO(111)表面结构模拟CuO表面释氧过程,首先对CuO(111)表面O2分子的形成与脱附过程进行模拟计算。
如图1c,CuO(111)表面O原子分为四配位O和三配位O,Cu原子分为四配位Cu和三配位Cu。当相邻两个三配位 Cu(图 3-IS)中一个变为一配位Cu时(一个三配位Cu失去两个相邻表面O原子),即变为类似Cu2O(111)表面的结构(图3-FS)。因此本文探索三配位Cu两端O原子形成O2分子过程,以模拟CuO→Cu2O+O2反应。结构优化后CuO(111)表面设为反应初始结构 IS;去除 IS中一个三配位Cu两端的O原子,将一个自由O2分子置于两个O空位连线的正上方6 Å处,并使O—O键与O空位连线平行,对系统进行结构优化即得到FS结构。移动一个三配位O原子使之接近另一个三配位 O原子,对新的结构进行DFT结构优化,发现两个O原子形成了稳定的O—O结构,即反应中间态结构,如图3-MS。对IS→MS和MS→FS进行过渡态搜索得到过渡态TS1和TS2,整个CuO表面释氧过程的结构变化如图3。
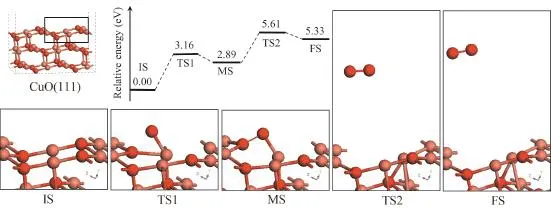
图3 CuO(111)表面释氧过程的结构及能量变化Fig. 3 The structures and the corresponding potential energy of oxygen release in the CuO(111)
由图3可看出,IS→MS过程中,三配位O首先与相邻的四配位Cu分离,迁移至四配位O上方;同时四配位O与下层Cu间的键断裂,向上位移并与三配位O成键,最终形成MS结构。MS中O—O键长为1.461 Å,大于自由O2分子键长1.225 Å;O—O键与表面的结合能为2.44 eV(Ea=E(FS)-E(MS)),在化学吸附热范围内。说明MS中形成了O2分子,且化学吸附于CuO表面。MS→FS过程中,吸附的 O2分子与表面 Cu原子之间的键断裂,并逐渐脱离表面,形成 FS结构。FS中 O2分子键长为1.248 Å,说明其为自由O2分子。因此,CuO表面释氧同样分为O2分子形成和O2分子脱附过程,与CuO团簇释氧过程类似。图 3同时给出了释氧过程的能量变化曲线,O2分子在CuO表面的形成势垒为3.16 eV,反应热为2.89 eV。与团簇中O2分子形成过程不同,CuO表面O2分子形成是吸热过程,且能量势垒较高,这主要是因为表面结构中O原子迁移需有多个Cu—O键断裂。O2分子的脱附势垒为 2.72 eV,小于第一步的 O2分子形成势垒,说明CuO表面释氧过程的限制步骤为O2分子形成过程。
2.3 氧原子在CuO(111)内的扩散过程
CuO内部O原子向表面的扩散性能是影响氧化铜释氧的另一个主要因素。为了探索影响CuO释氧的主要限制步骤,本文进一步计算了O原子在CuO内部的扩散势垒,追踪第二层O原子向表面层扩散的过程。由于内部O原子向表面扩散的过程相当于表面O空位向CuO内部O空位扩散,因此本文将CuO(111)平板模型的表面层去除一个O原子O3f,对其进行结构优化,作为反应的初始态 IS(图4-IS),追踪位于O3f下方的内层O原子Osub的扩散过程。去除CuO(111)的Osub,对其进行结构优化得到FS(图4-FS)。建立IS→FS的反应路径,进行过渡态搜索,得出结构及能量变化过程。
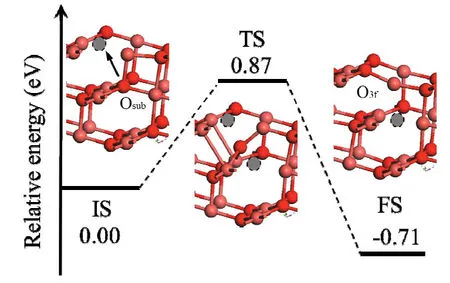
图4 CuO(111)内O原子扩散过程,灰色虚拟原子代表氧空位Fig. 4 O anion diffusion in the CuO slab,gray spot represents oxygen vacancy
从图4可看出,内层Osub首先迁移到第二层O原子与表面O原子间的空隙位置,并与下层Cu原子之间的键断开;Osub继续向表面迁移,逐渐与表层的Cu原子成键,直至最终结构FS。O原子扩散过程需克服的能量势垒为 0.87 eV,反应热为-0.71 eV。内层O原子向表面扩散为放热反应,且扩散势垒较低,表明反应易于进行。由计算结果可知,在CuO释氧过程中,O原子在CuO(111)内的扩散势垒(0.87 eV)显著小于O2分子在CuO(111)表面的形成势垒(3.16 eV)及脱附势垒(2.72 eV)。
以上结果表明,不论是团簇还是表面结构,CuO的释氧过程均需经过O2分子形成及O2分子脱附过程。与表面释氧过程相比,团簇释氧的 O2分子形成势垒很低,主要是由于表面结构Cu—O键排列更致密,O原子迁移需更多的键断裂。但团簇释氧的O2分子形成势垒相对较高(3.51 eV)。此外,虽然O原子向表面扩散势垒相对较低(0.87 eV),但随着氧载体不断释氧,CuO结构会发生改变,内部O原子的扩散仍有可能变为释氧限制步骤。
实验测得纯氧化铜的释氧活化能为 327 kJ/mol(3.39 eV)[11],这与团簇、表面释氧限制步骤的能量势垒(3.51 eV和3.16 eV)很接近。值得注意的是,实验得到的活化能反映的是整个释氧过程的表观活化能,包含了 O2分子形成、脱附,O原子扩散,氧载体结构变化等微观过程。通过DFT计算有助于理解释氧各个阶段的变化信息,为选择合适的氧载体提供理论依据。通过分析可知,O2分子形成或脱附的首要步骤是某些Cu—O键的断裂,因此,破坏O原子与周围Cu原子的成键是增强CuO释氧能力的关键。在此理论的基础上,通过合理改变CuO结构,如通过增加O缺陷、添加惰性负载或掺杂其他元素来改变表面结构和成键性质,以降低相应的能量势垒,可有效提高CuO的释氧性能,也为开发新型高效氧载体提供了一个有效途径。
3 结 论
本文基于 DFT理论的量子化学计算方法,对Cu4O4团簇模型和 CuO(111)周期性表面平板模型的释氧过程进行了模拟及计算,具体探索了Cu4O4→Cu4O2+O2过程中团簇结构的变化,以及反应不同阶段克服的能量势垒;研究了CuO(111)表面的O2分子形成、脱附过程和O原子在CuO(111)内部的扩散性能,得出以下结论:
(1)Cu4O4释氧需经过O2分子形成、脱附过程,且O2分子脱附是团簇释氧的限制步骤,其能量势垒为3.51 eV。
(2)CuO(111)表面氧脱附与Cu4O4团簇的释氧过程类似,均需经过O2分子形成和O2分子脱附两个过程,但CuO(111)表面O2分子形成势垒(3.16 eV)显著高于CuO团簇,主要是因为CuO表面的O原子与周围多个原子成键,结构更致密,O原子迁移困难。因此提高CuO表面释氧性能的关键是破坏表面O原子与周围Cu原子的成键。
(3)O原子由CuO(111)内部扩散至表面所需的能量势垒为0.87 eV,小于CuO(111)表面O2分子形成的势垒,说明 CuO表面释氧的限制步骤是表面O2分子形成过程。
(4)计算结果与已有实验值符合较好,说明量子化学计算可用于解释实验现象,并探索反应各个阶段的微观机理,有利于更好地理解化学反应过程的限制步骤,对提高和改善氧载体性能及开发新型释氧材料具有积极的指导意义。
[1] MATTISSON T,LYNGFELT A,LEION H. Chemicallooping with oxygen uncoupling for combustion of solid fuels[J]. International journal of greenhouse gas control,2009,3(1): 11-19. DOI: 10.1016/j.ijggc.2008.06.002.
[2] MATTISSON T,LEION H,LYNGFELT A. Chemicallooping with oxygen uncoupling using CuO/ZrO2with petroleum coke[J]. Fuel,2009,88(4): 683-690. DOI: 10.1016/j.fuel.2008.09.016.
[3] ADÁNEZ-RUBIO I,ABAD A,GAYÁN P,et al. Performance of CLOU process in the combustion of different types of coal with CO2capture[J]. International journal of greenhouse gas control,2013,12: 430-440. DOI: 10.1016/j.ijggc.2012.11.025.
[4] SIRIWARDANE R,TIAN H J,MILLER D,et al. Evaluation of reaction mechanism of coal-metal oxide interactions in chemical-looping combustion[J]. Combustion and flame,2010,157(11): 2198-2208. DOI:10.1016/j.combustflame.2010.06.008.
[5] DONG C Q,LIU X L,QIN W,et al. Deep reduction behavior of iron oxide and its effect on direct CO oxidation[J]. Applied surface science,2012,258(7): 2562-2569. DOI:10.1016/j.apsusc.2011.10.092.
[6] TAN Q L,QIN W,CHEN Q L,et al. Synergetic effect of ZrO2on the oxidation-reduction reaction of Fe2O3during chemical looping combustion[J]. Applied surface science,2012,258(24): 10022-10027. DOI: 10.1016/j.apsusc. 2012.06.067.
[7] LI F X,LUO S W,SUN Z C,et al. Role of metal oxide support in redox reactions of iron oxide for chemical looping applications: experiments and density functional theory calculations[J]. Energy & environmental science,2011,4(9): 3661-3667. DOI: 10.1039/C1EE01325D.
[8] PERDEW J P,BURKE K,ERNZERHOF M. Genera-lized gradient approximation made simple[J]. Physical review letters,1996,77(18): 3865-3868. DOI: 10.1103/ PhysRevLett.77.3865.
[9] Delley B. Analytic energy derivatives in the numerical local-density-functional approach[J]. The journal of chemical physics,1991,94(11): 7245-7250. DOI: 10.1063/1.460208.
[10] 张健. 镁及其合金氢化物吸放氢性能及电子机制研究[D]. 长沙: 湖南大学,2009.
[11] Sahir A H,Sohn H Y,Leion H,et al. Rate analysis of Chemical-Looping with Oxygen Uncoupling (CLOU) for solid fuels[J]. Energy & fuels,2012,26(7): 4395-4404. DOI: 10.1021/ef300452p.
Oxygen Release Mechanisms of Cu-Based Oxygen Carriers Based on Density Functional Theory Calculations
FENG Xiao-ming,FENG Yong-xin,LIU Ya-ming,LI Fang-yong
(Electric Power Research Institute of Guangdong Power Grid Company,Guangzhou 510080,China)
Chemical-looping with oxygen uncoupling (CLOU) is a new combustion method based on chemical-looping combustion technology,which allows intrinsic separation of pure CO2from hydrocarbon combustion. The key point of CLOU is finding an ideal oxygen carrier that has good oxygen adsorption and releasing characters. The reactivity of oxygen carriers in macro-level is determined by the mechanism of microscopic lattice oxygen transportation,but few attentions have been paid on the subject currently. This article used density functional theory (DFT) to study the oxygen release mechanism of CuO carriers,the CuO clusters and slab models were built to simulate the process of cluster and surface oxygen release. CuO slab model’s oxygen release process includes inward oxygen atom diffusion,the formation of O2molecule in surface and the release of O2molecule. The results indicate that the highest energy barrier during the CuO(111) surface oxygen release is 3.16 eV,which is close to the experiment value 3.39 eV,but lower than that of CuO cluster model. And besides,the energy barrier of oxygen atom diffusion inside the CuO(111) is just 0.87 eV,indicating that the surface oxygen molecules’ formation process is the rate-limit step of CuO(111) oxygen release.
density functional theory (DFT);chemical-looping with oxygen uncoupling (CLOU);Cu-based oxygen carriers;oxygen release mechanisms
TK16
A
10.3969/j.issn.2095-560X.2016.05.009
2095-560X(2016)05-0393-06
冯晓鸣(1985-),男,硕士,工程师,主要从事烟气脱硫脱硝的研究。
冯永新(1968-),男,博士,高级工程师,主要从事电力设备监测及诊断的研究。
刘亚明(1979-),男,博士,高级工程师,主要从事电力行业节能减排的研究。
李方勇(1984-),男,硕士,高级工程师,主要从事燃煤机组调试方面的研究。
2016-06-08
2016-08-17
† 通信作者:刘亚明,E-mail:dbqpun@foxmail.com
猜你喜欢
华人时刊(2022年9期)2022-09-06
大学物理(2022年1期)2022-01-13
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
华人时刊(2020年15期)2020-12-14
电子制作(2017年19期)2017-02-02
郑州大学学报(理学版)(2014年4期)2014-03-01
中国火炬(2013年11期)2013-07-25
中国火炬(2013年10期)2013-07-24
