
用于合成聚醚多元醇的催化剂研究进展
2016-10-21 01:58:06赵瑞超姚丽娜盖东杰
上海塑料 2016年1期
赵瑞超,姚丽娜,盖东杰,顾 尧
(青岛科技大学 橡塑材料与工程教育部重点实验室, 山东 青岛 266042)
用于合成聚醚多元醇的催化剂研究进展
赵瑞超,姚丽娜,盖东杰,顾尧
(青岛科技大学 橡塑材料与工程教育部重点实验室, 山东 青岛 266042)
综述了阴离子、阳离子、双金属氰化物配位催化剂和新型磷腈催化剂催化环氧化合物的聚合机制,并详细介绍了磷腈催化剂的制备方法及发展趋势。
阳离子聚合; 阴离子聚合; 双金属氰化物配位催化剂; 磷腈催化剂
0 前言
聚醚多元醇作为聚氨酯的主要原料,它的性能将直接影响最终产物的性能。醚键是一种特征性的碱性强键,所以环醚的开环聚合反应只能用阳离子型引发剂引发[1]。但是,三元环氧化物是个例外。由于其环的变形程度大,环的张力能及聚合热也大,聚合自由焓的负性大,因此,环的稳定性低,容易开环聚合,再加上—C—O—键是极性键,富含有电子的氧原子易受阳离子进攻,缺电子的碳原子容易受到阴离子的进攻。在动力学上,三元环醚也极易聚合,故可以使用阳离子开环聚合、阴离子开环聚合和配位聚合。传统的阴离子聚合和阳离子聚合存在产物的相对分子质量较低、相对分子质量分布较宽及不饱和度高等缺点。双金属氰化物配位催化剂(DMC)的使用可以追溯到20世纪60年代,当时美国的GEN TIRE & RUBBER公司[2-3]首次开发了双金属氰化物配位催化剂[4],并应用于环氧丙烷的开环聚合反应,合成了具有较高相对分子质量、较低不饱和度的聚醚多元醇。此后,诸多专家学者在研究制备DMC催化剂的同时,也对其催化聚合的机制进行了深入的探讨[5~10]。DMC催化聚合环氧化合物的反应机制属于配位聚合,其活性中心是螯合的金属离子。这种催化剂虽然对阴离子、阳离子聚合进行了改进,但又产生了新的问题,如不能使用小分子起始剂,聚醚分子的头~尾的聚合选择率低,以及不能用环氧乙烷直接封端等[11]。DMC催化剂在环氧丙烷加成反应中具有很高的催化活性,在环氧乙烷加成反应中,链增长速率大于链转移速率,使得聚醚多元醇中有的聚氧化乙烯支链很长,有的链段很短,甚至没有,导致分子链的长短不均,产物的相对分子质量分布宽[12]。笔者介绍了阳离子(质子酸、路易斯酸)、阴离子(路易斯碱)、双金属氰化物配位催化剂及磷腈催化剂催化环氧化合物的聚合机制[13],并详细介绍了磷腈类催化剂的合成方法及发展趋势。
1 阳离子开环聚合[14]
使用阳离子催化剂催化环氧化合物开环聚合,符合活性聚合的特性,如活性种寿命长、相对分子质量分布窄、引发速率比增长速率快等,具有快引发、慢增长等特征。在聚合过程中,酸根离子可以与环氧化合物单体争夺质子,并伴有链转移和解聚反应,最终的产物只有相对分子质量很低的聚合物。其次,阳离子聚合反应产物的相对分子质量分布较窄,反应条件较为苛刻,副反应也较多,除了生成聚乙二醇和聚丙二醇外,还生成具有强致癌性的二鎓烷和2-甲基鎓烷等低分子齐聚物。另外,酸腐蚀设备严重,因此工业上很少应用。
阳离子型引发剂包括质子酸和路易斯酸。质子酸有浓硫酸、磷酸、氯磺酸(HSO3Cl)、氟磺酸(HSO3F)、三氯代乙酸(CCl3COOH)等。这些强质子酸在非水介质中能部分电离,产生H+,进而引发聚合。但酸中阴离子的亲核性不能太强,如氢卤酸,以免与质子或阳离子共价结合而反应终止。路易斯酸有BF3、AlCl3、TiCl4、SnCl4等。但是路易斯酸引发活性低,需要添加微量共引发剂作为阳离子源,才能保证正常聚合。一般引发剂与共引发剂有一最佳比,才能获得最大聚合速率和最高相对分子质量。定性地说,如果共引发剂少,则活性不足;共引发剂过多,则反应将终止。例如:水作为质子供体时,水量过多就会使聚合活性降低。原因有:(1) 生成活性较低的氧鎓离子;(2) 向水转移,产生无活性的“络合物”。
1.1质子酸催化环氧丙烷开环聚合机制
1.1.1链引发和链增长
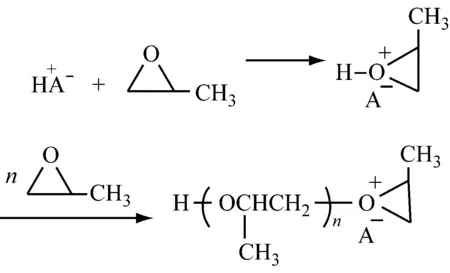
1.1.2链转移
1.1.2.1向其他分子链转移
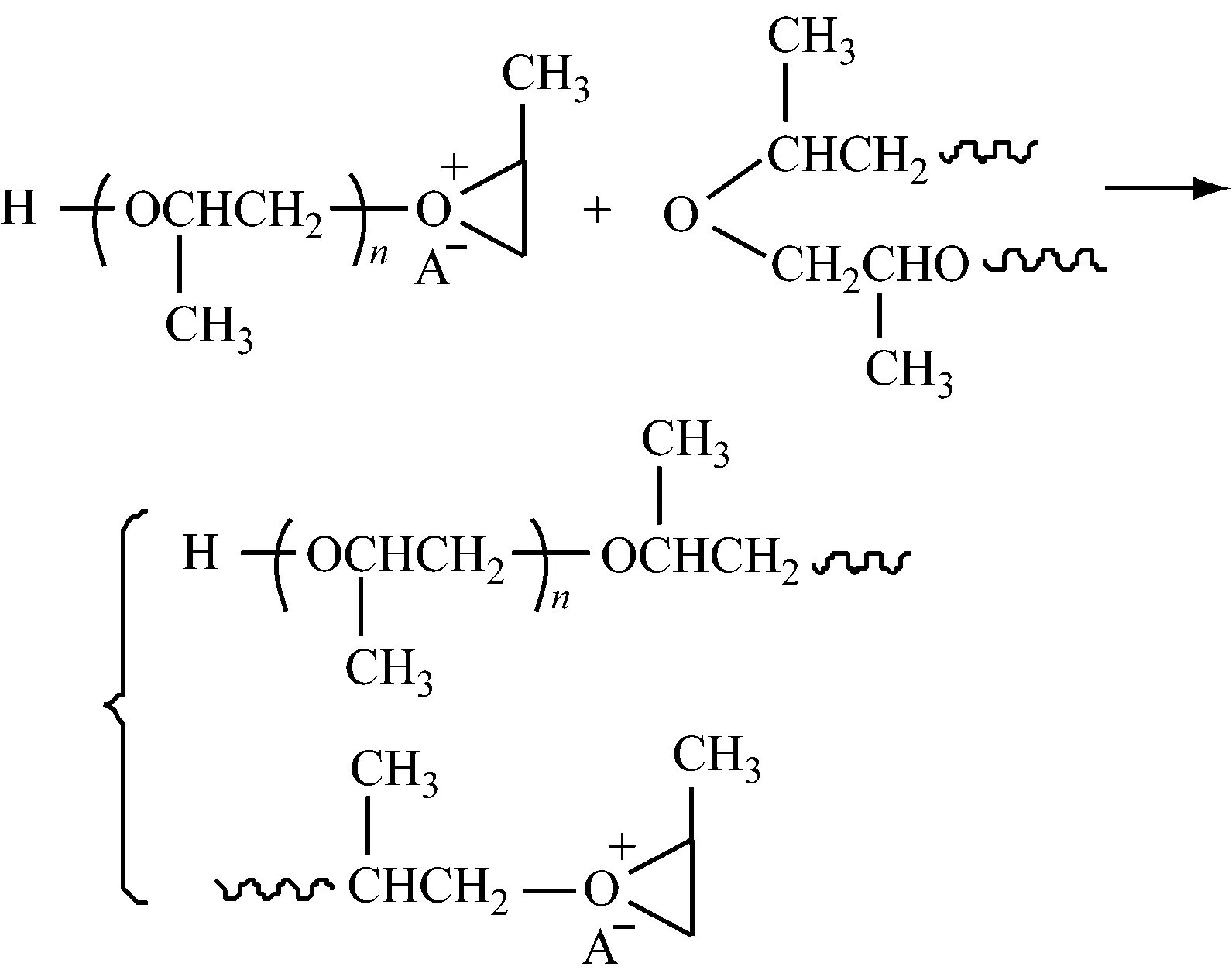
1.1.2.2向单体转移
1.1.3链终止
(1) 当反离子的亲和性足够强时,将与增长链末端的碳阳离子结合,使链增长终止;(2) 活性中心与反离子的一部分结合,使链增长终止;(3) 人为添加一些低活性的物质,使链增长终止。
ACH2CH2OCH2CH2OH+BX
注:HX可以是水、醇、酸等
1.2路易斯酸催化环氧丙烷开环聚合机制
1.2.1活性中心的生成
路易斯酸的反应机制比质子酸的多了一步活性中心的生成,链引发、链增长和链转移的过程与质子酸的反应机制相同。
R为H、CH3O、C2H5O
1.2.2链引发、链增长和链转移
链引发、链增长和链转移的过程与质子酸的反应机制相同。
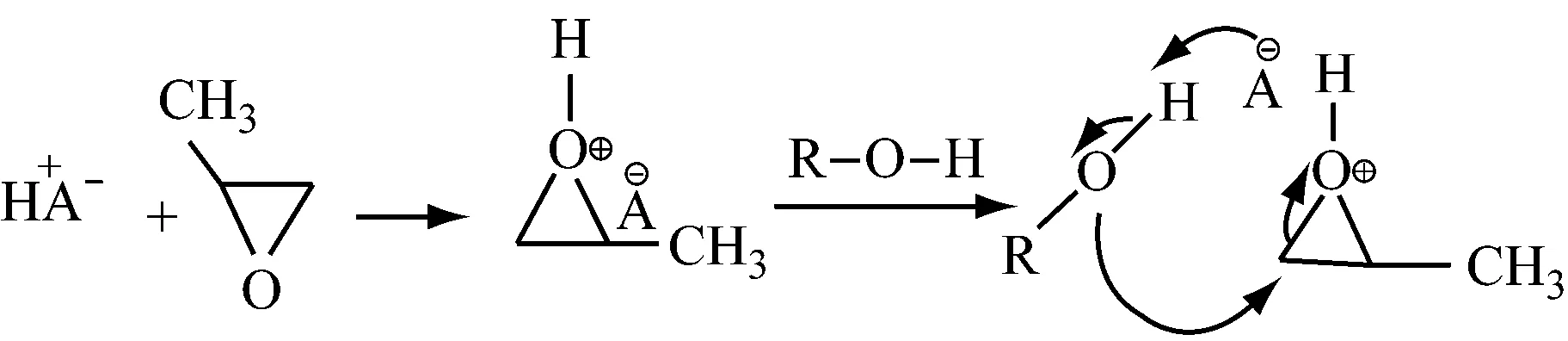
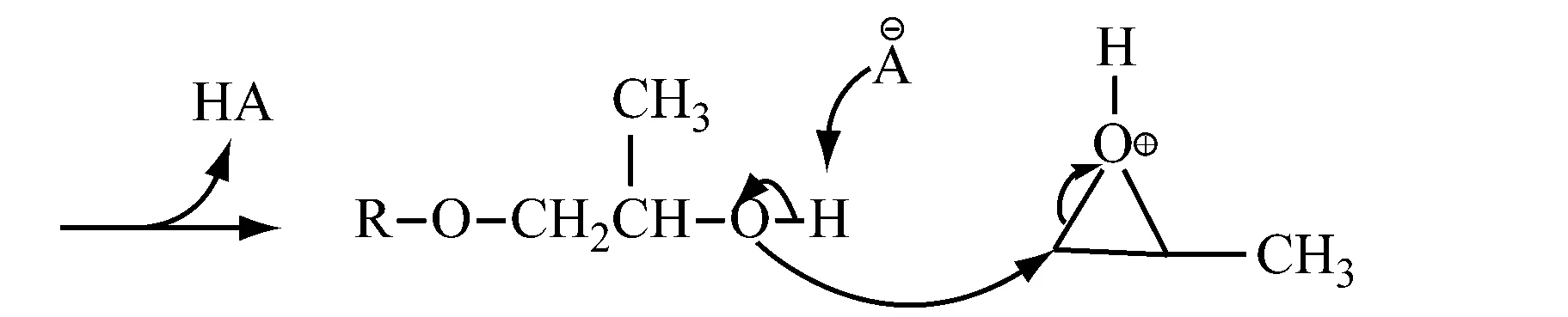
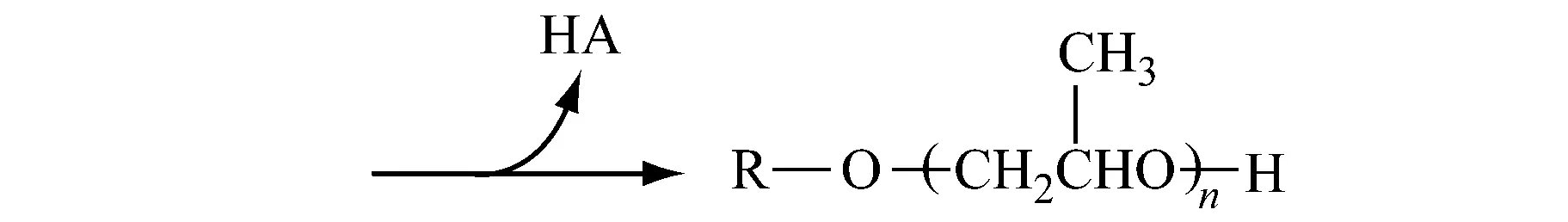
阳离子聚合反应中真正意义上的动力学链终止反应比较少,不像阴离子聚合那样无终止而成为活性聚合,阳离子聚合反应的副产物多,限制因素较多,因此很少在工业上应用。
2 阴离子开环聚合
环氧化物的阴离子聚合反应是以活泼氢化合物作为起始剂,环氧化物与碱金属氢氧化物或其醇盐作用,产生醇盐阴离子引发反应,而这种阴离子在增长阶段通过与单体分子连续开环反应生成链聚合物,反应条件比较温和,反应比较容易控制。
环氧烷烃开环聚合常用的阴离子引发剂有碱金属的烷氧化物(如醇钠)、氢氧化物、氨基化物、有机金属化合物、碱金属氧化物等。环氧乙烷是高活性单体,低活性甲醇钠就足以引发聚合。下面以氢氧化钾为例,说明环氧烷烃阴离子聚合的反应机制。
2.1氢氧化钾催化环氧丙烷开环聚合机制
2.1.1活性中心的生成
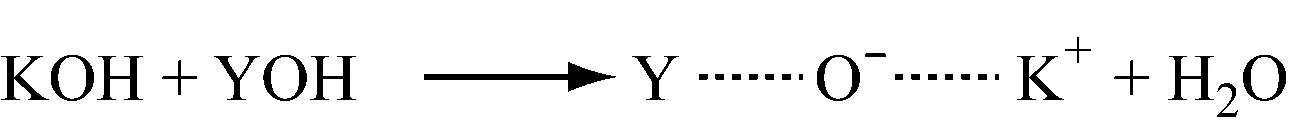
2.1.2链引发

2.1.3链增长
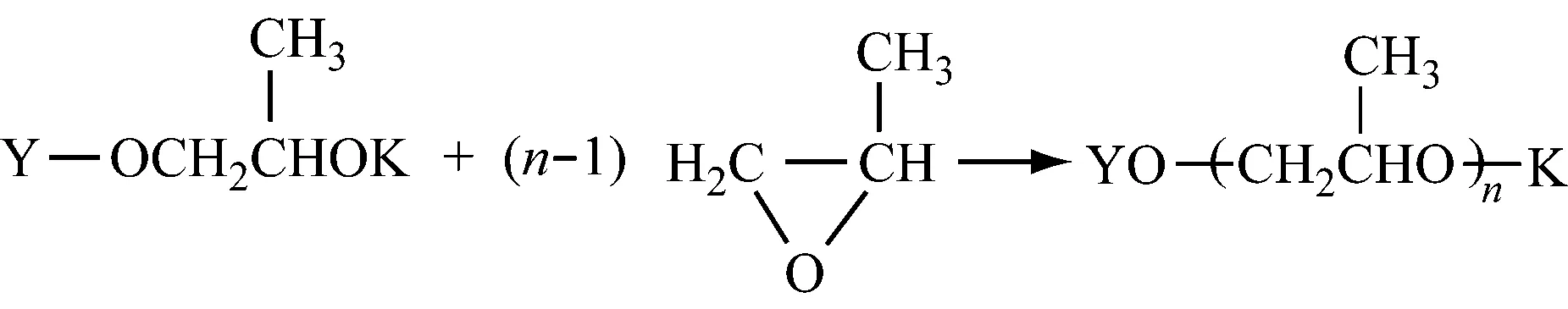
2.1.4链转移
2.1.4.1向起始剂转移
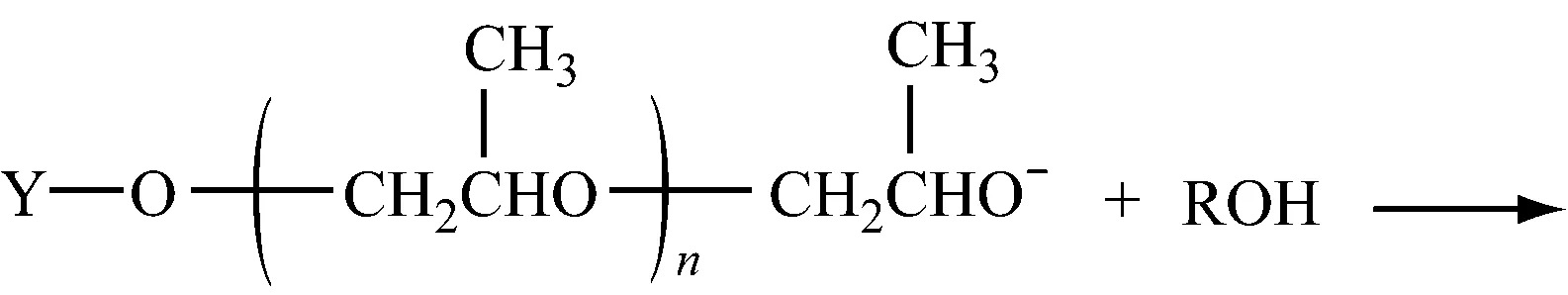
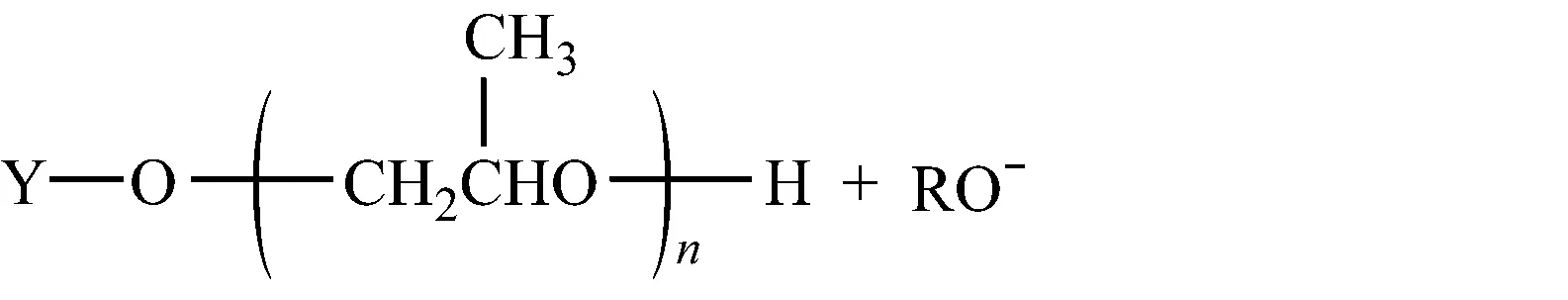
2.1.4.2向大分子转移
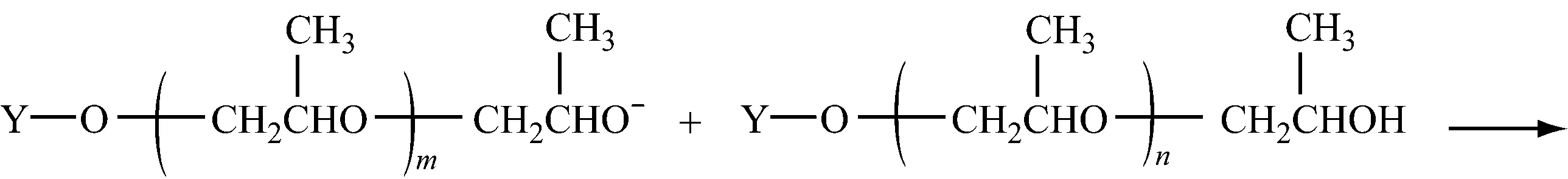
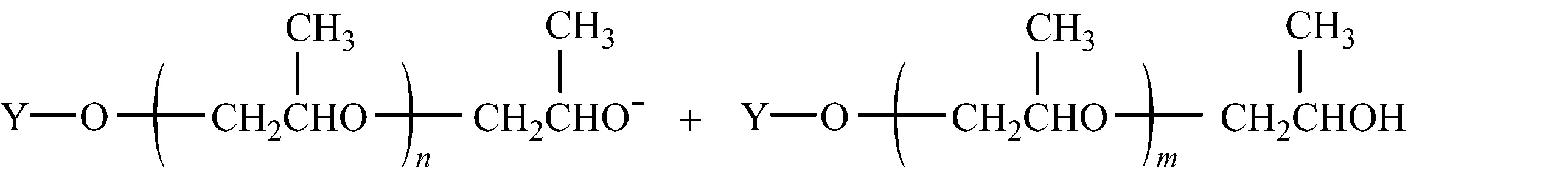
2.1.4.3向单体转移

2.1.5链终止
链终止属于活性阴离子聚合机制,即由引发和增长两步基元反应组成。想要结束反应,需要加入草酸、磷酸等质子酸,使活性链失活,进而控制产物的相对分子质量。

如果不加终止剂而加环氧乙烷,该反应则聚合成:(1) 两性嵌段共聚物,可以用作表面活性剂;(2) 用环氧乙烷封端的聚氧化丙烯,可以作为聚氨酯的原材料,提高产物的力学性能。
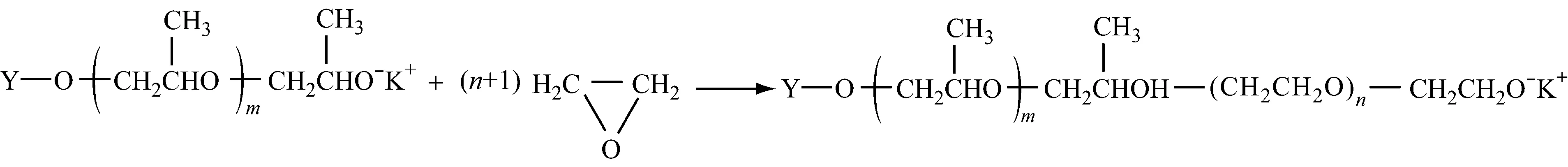
阴离子催化聚合环氧化物得到的聚合物的相对分子质量很低,其中催化环氧丙烷得到的聚合物相对分子质量最高不过7 000。这是因为环氧化物的阴离子催化活性较低,以及存在向单体的链转移反应。在聚合反应过程中存在着起始剂与阴离子增长链的快速质子交换反应[15],使聚合度降低,相对分子质量分布变宽。向单体的链转移反应,环氧丙烷尤为突出,环氧丙烷的甲基上的氢原子容易被夺取而转移,转移后形成的单体活性种又会很快转变成烯丙醇钠离子对,进而继续引发聚合。此外,产生的丙烯基或烯丙基端基会发生重排现象(不饱和端基的出现),这增加了体系中聚合物链的数目,也会使相对分子质量降低,相对分子质量分布变宽。Simons等[16]指出:烯丙基氧离子与丙烯基氧离子可以互相重排。例如:催化环氧乙烷时,不存在不饱和度增加,而催化环氧丙烷时,不饱和度增加。这说明环氧丙烷发生重排反应产生双键。Gee等[17]认为:链转移是另一种重排反应,重排后的产物可以继续引发聚合,这也是产生副产物的原因。
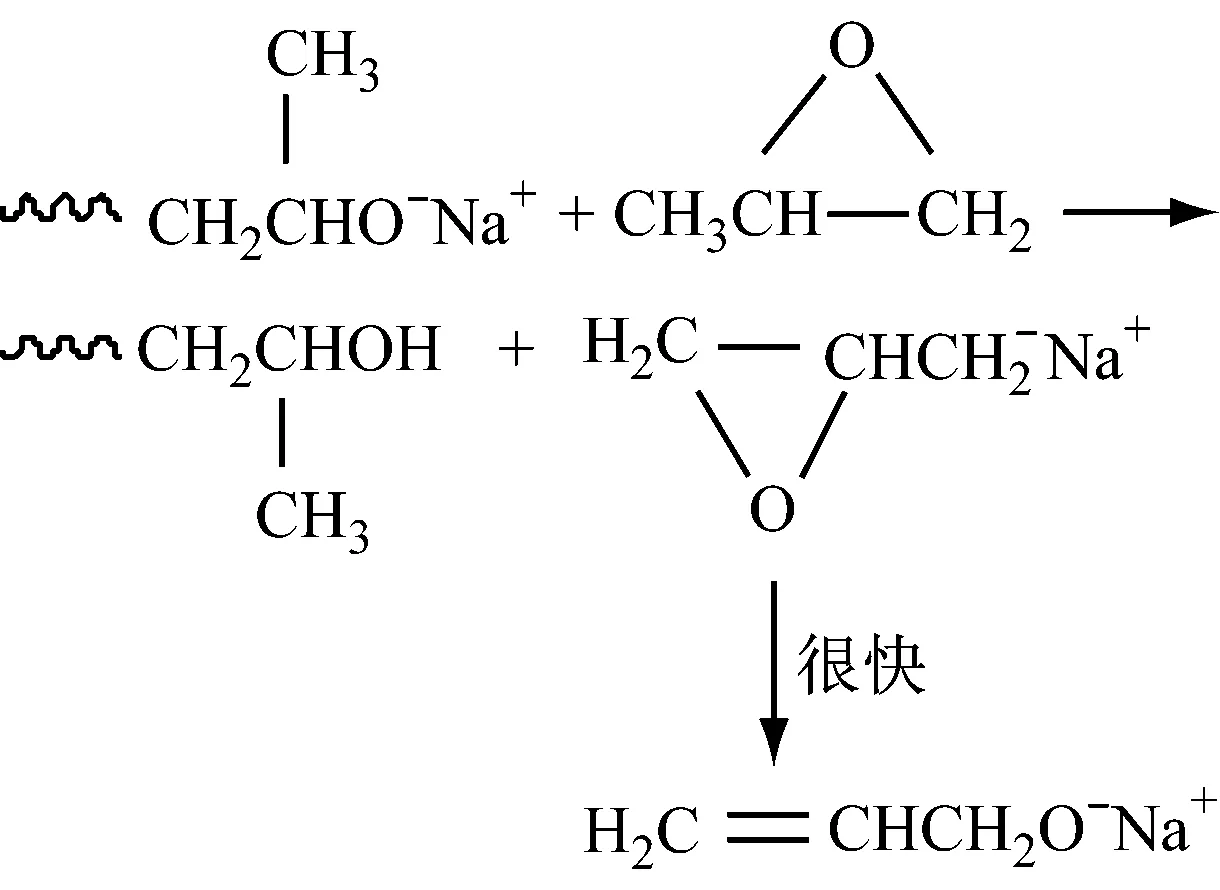
另外,聚合反应结束后,必须从聚醚产物中将催化剂残留物分离出来,否则会导致下游产品发生不必要的副反应。聚醚的精制工艺比较复杂,一般包括中和、吸附、脱色、脱水和压滤等步骤[18]。
3 双金属氰化物配位催化剂
双金属氰化物配位催化剂(DMC),是催化环氧丙烷、环氧乙烷等环氧化合物开环聚合,合成高相对分子质量、低不饱和度的聚醚多元醇的高效配位催化剂。DMC催化剂一般是由水溶性金属氰化物配合物和金属盐水溶液,在有机配体的作用下反应制得的。DMC催化剂具有结构空位[19],并可以形成配位通路的正八面体,它是由Co(CN)63-、Zn2+、叔丙醇(TBA)、聚丙二醇(PPG)、H2O等配位而成的。非晶态结构的DMC催化剂[20]具有潜在的高催化活性,而在制备DMC催化剂时,体系中有机配体的存在有利于形成非晶态结构。DMC催化剂中的配体可以用水溶性小分子有机物,加入有机配体后能提高活性,但是配体的类型会影响催化剂的活性。DMC催化合成的聚醚产物的相对分子质量可由转化单体与起始剂的量之比控制[21],而与催化剂没有直接关系。另外,分批加料可以获得相对分子质量分布较窄的聚合物;一次加料则得到相对分子质量分布较宽的聚合物。这是因为一次加料浓度较大,单体浓度不均匀。
用双金属氰化物作为催化剂,合成的聚醚具有以下优点[22]:
(1) 调控产品的相对分子质量通过起始剂参与的链转移反应来调节产品的相对分子质量。控制起始剂与单体的量之比,可合成相对分子质量从几百至几万的聚醚多元醇,且合成的聚醚多元醇的最高相对分子质量比用KOH催化剂制得的高;这里的起始剂仅在链转移阶段起作用,它并不是链引发和链增长阶段必需的成分。
(2) DMC催化剂的反应活性高DMC催化剂使用的量远比KOH的少,而催化活性是KOH的50~150倍。
(3) 聚醚多元醇的相对分子质量分布窄Mw/Mn<1.4。这是由于在聚合反应中,活化的催化剂和含有羟基的化合物的配位速率大大高于链增长的速率和单体异构化的速率,弥补了阴离子聚合过程中通过质子转移使聚合物增长的缺陷,环氧化合物的转化率得以提高,又使环氧丙烷异构化的几率降低,致使最终产品的相对分子质量分布较窄。
但同时又产生了新的问题,如引入了金属离子,对产物的纯度以及后续的提纯加工产生了影响;高质量的DMC催化剂少,且成本高;聚合过程中起始剂仍要使用KOH碱催化剂制得的低相对分子质量的聚醚。小分子起始剂仍不能实现工业化,原因是小分子起始剂一般有较高的链转移速率,采用起始剂先加入法时[23],聚合速率明显降低,甚至反应很快停止。在生产中可以采用起始剂后加入法,一方面,可以有效地控制产物的相对分子质量;另一方面,可以降低反应釜中反应物的黏度,增加分子间的碰撞几率,提高原料转化率。最后,在聚合环氧丙烷时,不能用环氧乙烷直接封端。这是因为两者的单体竞聚率不同,在聚合过程中,两种单体环氧乙烷和环氧丙烷的聚合反应活性相差较大,环氧乙烷的活性远高于环氧丙烷的。当用环氧乙烷封端时,由于DMC催化活性高,得到的共聚物是以环氧乙烷聚合为主的链段,以环氧丙烷聚合为主的链段,以及少部分环氧乙烷封端的链段。
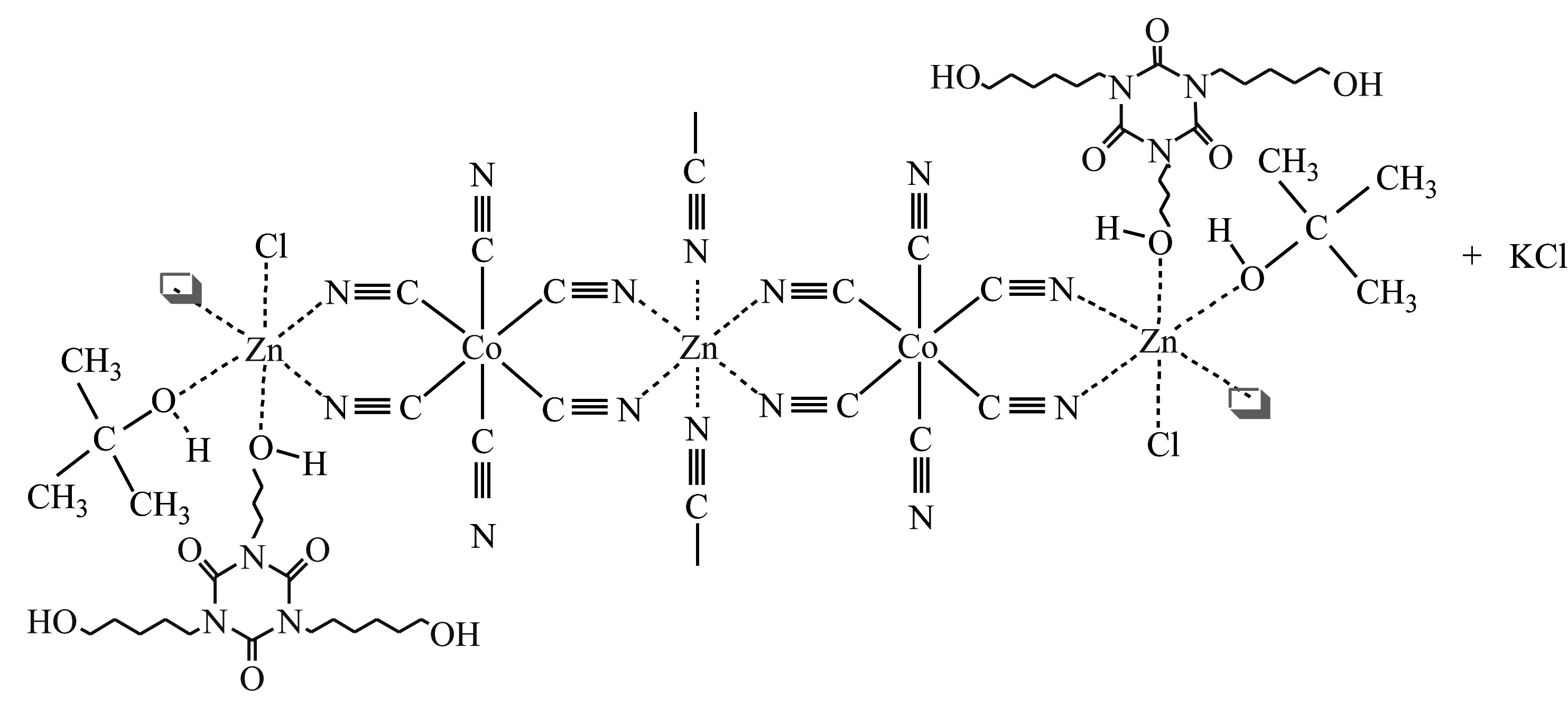
通过原位聚合方法[24]制备乳液状超微细DMC催化剂(NEDMC),简化了DMC催化剂的制备工艺[25]。NEDMC是一种高活性的DMC催化剂,而且便于储存和使用。
合成EDMC的技术路线:

DMC催化PO开环聚合的机制如下:
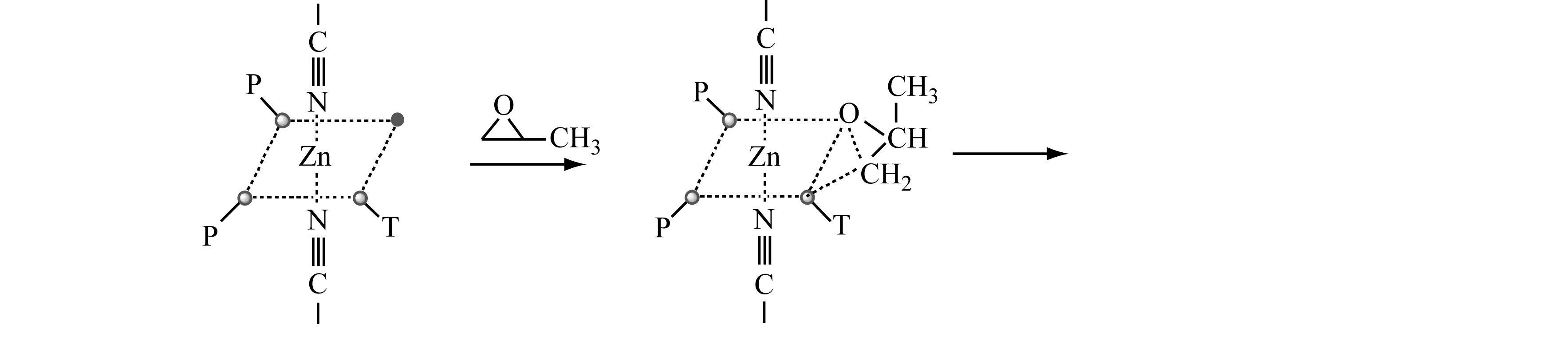
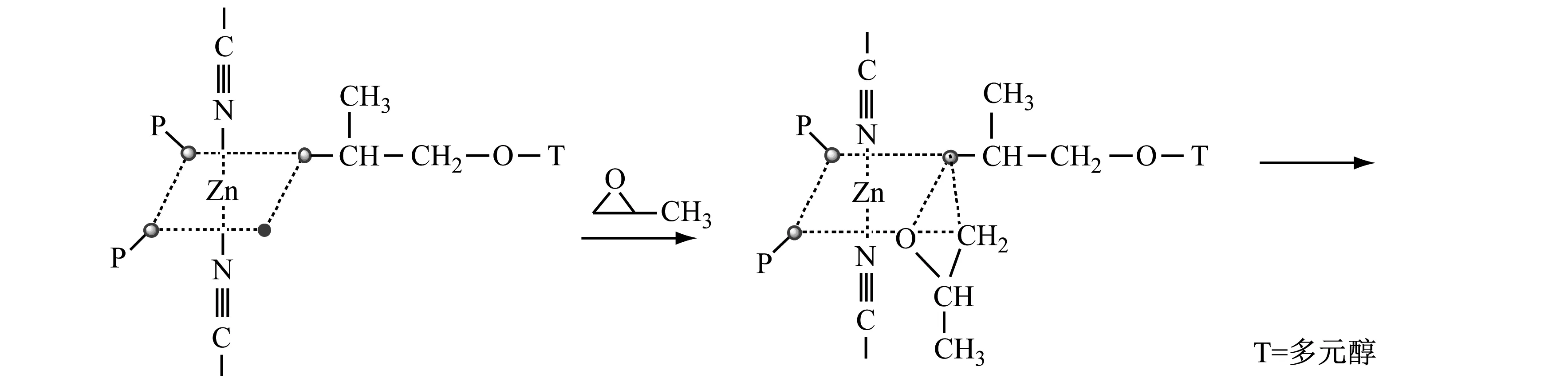
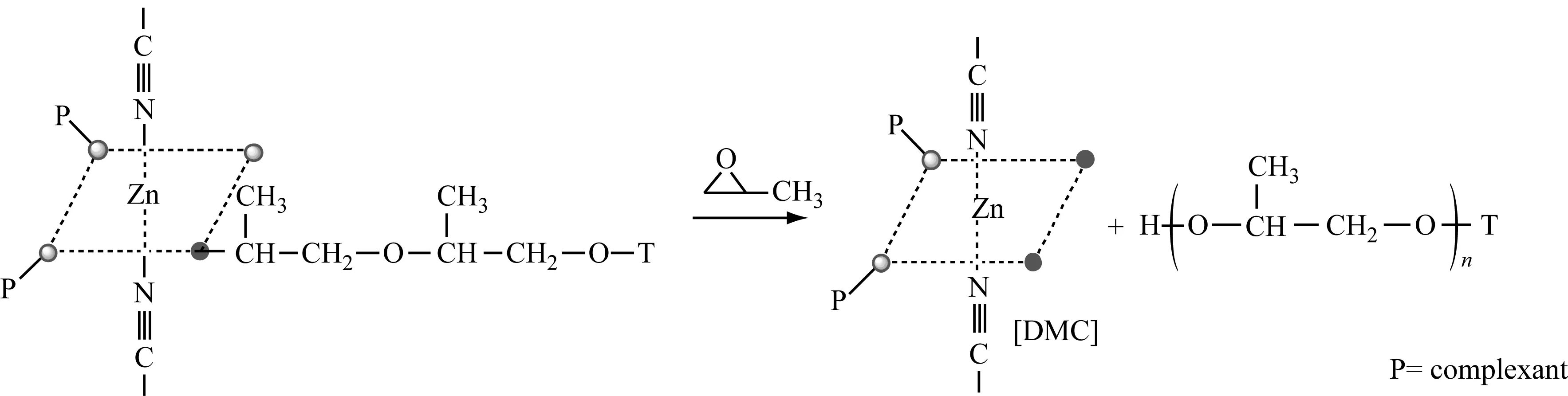
由上述可知:无论是无机催化剂还是有机金属催化剂都有其优缺点。为了最大化利用催化剂的优点,开发了新型的有机催化剂——磷腈类催化剂[26]。
4 磷腈类催化剂
在很多有机阴离子反应中,如阴离子亲核取代反应,作为反离子的阳离子对反应的活性和选择性起相当大的作用,所以可以通过修饰阳离子的空间结构[27]和电性能来提高阴离子反应的活性和选择性。例如:为了增强阴离子反应的活性和选择性,用冠醚取代碱金属作为阳离子。原因是阴离子活性种和阳离子隔离得较好,由于位阻效应和电子效应,阴离子聚合的活性就较高。在设计反离子结构时,可以认为反离子就是系统中允许阳离子通过的通道,这样阳离子就高度离域化[28],以稳定过渡态结构使阳离子通道结构具有有效的离子分离系统[29]来获得高度离域的阳离子。而磷腈类催化剂的阳离子就符合上述条件,所以磷腈盐和氧化磷腈催化剂提高了聚合反应活性,降低了副反应,合成了高相对分子质量的聚醚。为了获得高活性的磷腈催化剂,必须符合以下条件[30]:(1) 反离子与阴离子有很好的电子隔离;(2) 反离子必须高度离域化以获得稳定的过渡态结构;(3) 反离子具有疏水的表面以减少和阴离子活性种的表面反应;(4) 为了防止阴离子活性种进入反离子内部,设计的反离子最好具有闭环结构。
磷腈类催化剂不仅符合上述条件[31],即分子尺寸足够大;分子中阳离子的电荷是离域化的;含有单一的活性点,而且不含金属元素,避免了杂质的产生;分子对称排列,有利于提高其稳定性。磷腈类催化剂具有很好的催化效果,制得的聚醚不饱和度低、相对分子质量高、相对分子质量分布窄,还
可以直接用环氧乙烷封端,并可使用小分子作为起始剂等优点。利用磷腈类催化剂合成聚醚多元醇,制备的高回弹软泡的回弹率达到81%;而传统聚醚制备的只有50%~70%,而且制备的泡沫塑料的质量轻20%,这就增加了汽车坐椅的耐久性和乘坐的舒适性[32]。这对简化国内现有的生产工艺,降低能耗物耗,缩短生产周期,减少“三废”排放,提高产品的性能等有着重要意义。
磷腈又叫磷氮烯(phosphazene),是一类非金属的含有P=N双键的强碱性化合物,包括磷腈、磷腈盐和氧化磷腈。磷腈催化剂呈强碱性,如果存在于聚醚中会严重影响聚醚的性能和稳定性,因此必须除去。通常采用特定组成的吸附剂吸附,还可以用中和-吸附或萃取法工艺去除,可使催化剂的质量分数降至50×10-6以下。磷腈催化剂价格昂贵,需要回收利用,可以将预先制成的磷腈盐连接到载体上,通过过滤、洗涤、干燥后反复使用。
4.1磷腈类催化剂的催化机制
磷腈类催化剂在聚合过程中,活性中心都是以阳离子的形式存在,催化环氧丙烷阴离子开环聚合,聚合后残留的催化剂用阳离子交换树脂去除[33]。研究发现:将磷腈类催化剂去除干净并不好,在质量分数分别为85×10-6~10 000×10-6的磷腈阳离子与400×10-6~5 000×10-6的抗氧剂(如3,5-二叔丁基-4-羟基苯等)的协同作用下[34],才能获得贮存稳定性良好的聚醚多元醇,同时也简化了精制工艺。
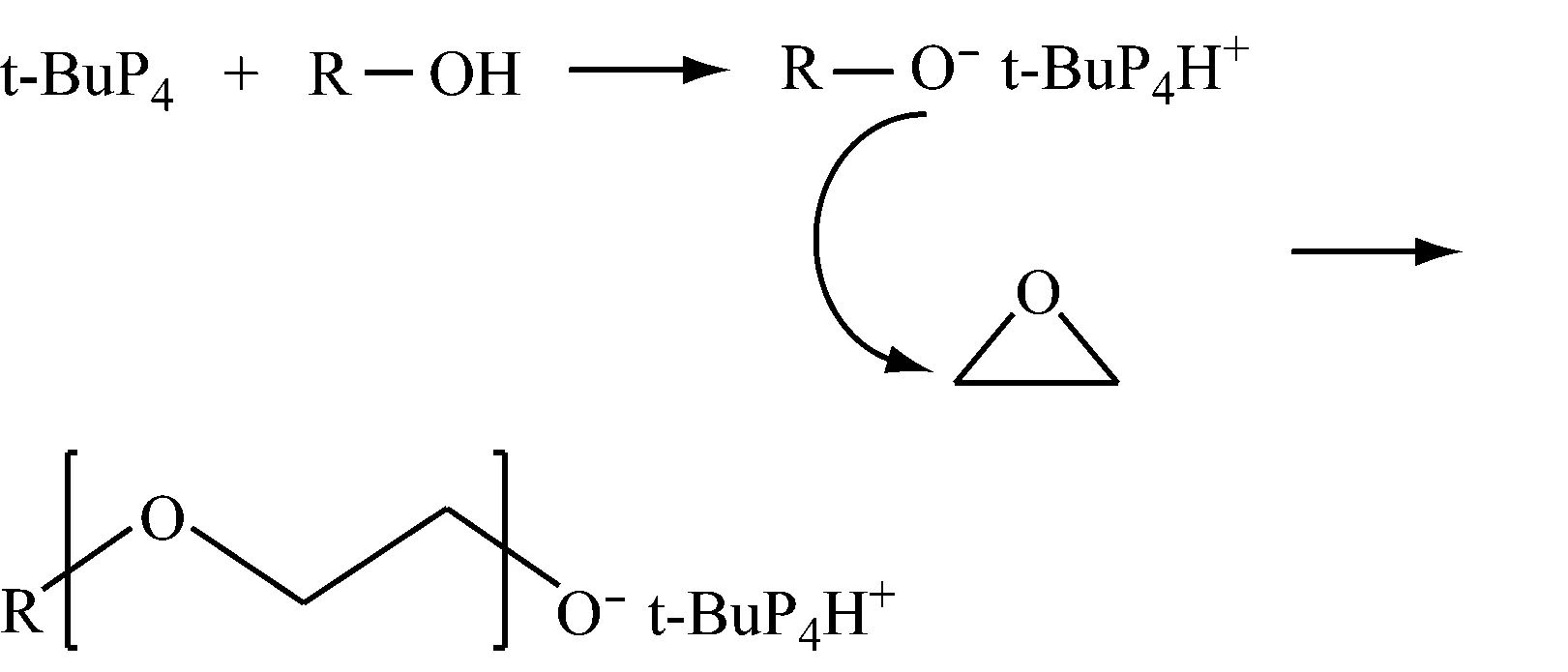
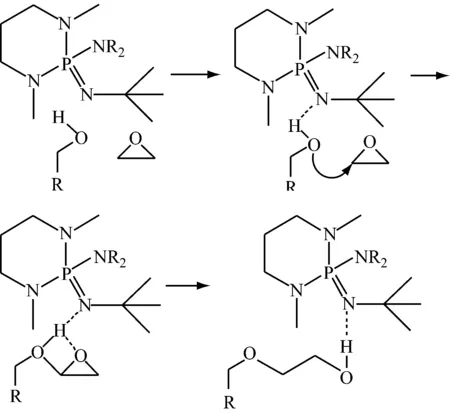
文献[35,36]表明:用甲醇为引发剂时,得到相对分子质量分布较窄的聚合物 (小于1.1),其摩尔质量与实测的(5 800 g/mol)接近。以正辛醇为引发剂时,其摩尔质量与实测的相差较大。这主要是因为正辛醇的对称性太低。另外,在研究引发体系的反应动力学时,发现反应并不符合一级动力学规律,随着反应时间的延长,一级动力学曲线的斜率逐渐增大。这可能是引发反应慢导致的。因为只有ROH被t-BuP4夺取质子后才能引发聚合反应。
4.2磷腈催化剂的制备
磷腈催化剂的制备分几步完成。首先,制备亚氨基正磷,其次,制备磷腈盐。由磷腈盐制备的氧化磷腈才是对环氧化物具有催化作用。
4.2.1亚氨基正磷的制备
(1) 氨基三(二甲氨基)氯化正膦的制备
现场配制PCl5。因为PCl5很难加入到反应器中,而且对水分很敏感,在储存时容易自粘结。在芳烃或卤代芳烃溶剂中,PCl3和Cl2反应生成PCl5,然后设定在76.1 ℃蒸出未反应的PCl3。水的质量分数<0.4%,在10~50 ℃,压力0.1~0.5 MPa,氮气保护下,n(二甲胺)∶n(PCl5)=(6.0~6.2)∶1.0,n(NH3)∶n(PCl5)=(2~20)∶1,不需要分离和提纯,直接加入二甲胺反应。然后再让其与氨反应制备氨基三(二甲氨基)氯化正膦。反应完成后,在加热条件下过滤除去不溶物,最后对滤液冷却结晶得到产物。该工艺简单,安全,原料利用率高。
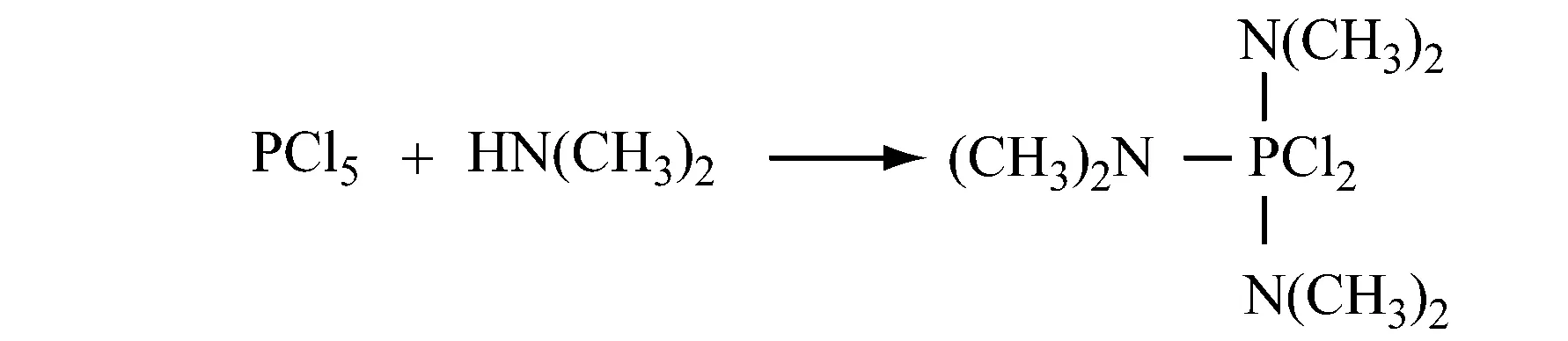
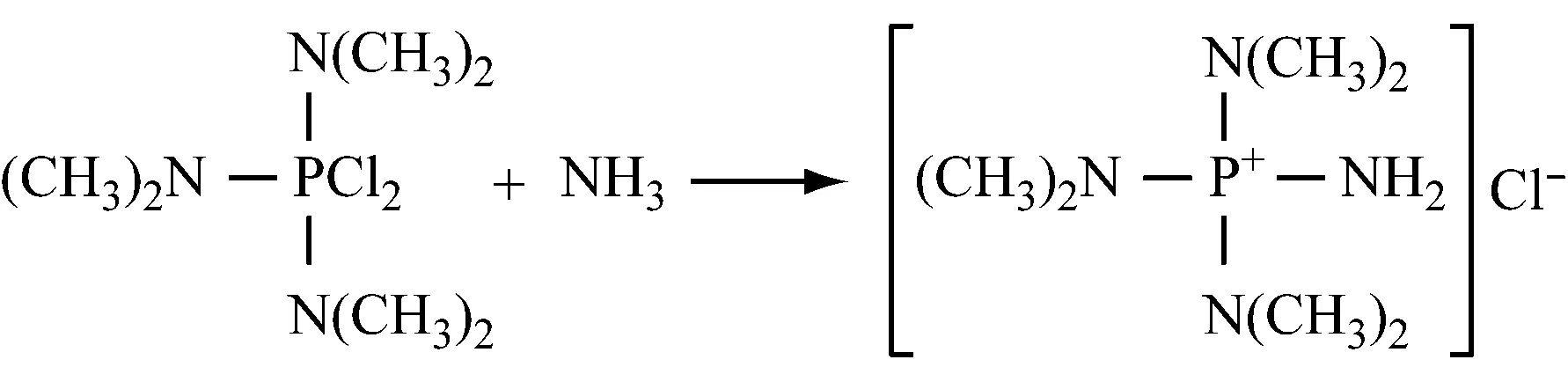
(2) 亚氨基三(二甲氨基)膦烷(正膦)的制备
在微量醇或水存在下,在卤代芳烃或烃类溶剂中,氨基三(二甲氨基)氯化正膦与碱金属氢氧化物或其醇盐反应制备亚氨基三(二甲氨基)膦烷。n(碱金属化合物)∶n[氨基(二甲氨基)氯化正膦]为(1~3)∶1,反应温度20~70 ℃,醇或水的质量分数为1.0%。反应完成后真空蒸馏,过滤去除不溶物,然后对母液蒸馏除去水和醇,再进一步蒸馏回收溶剂。
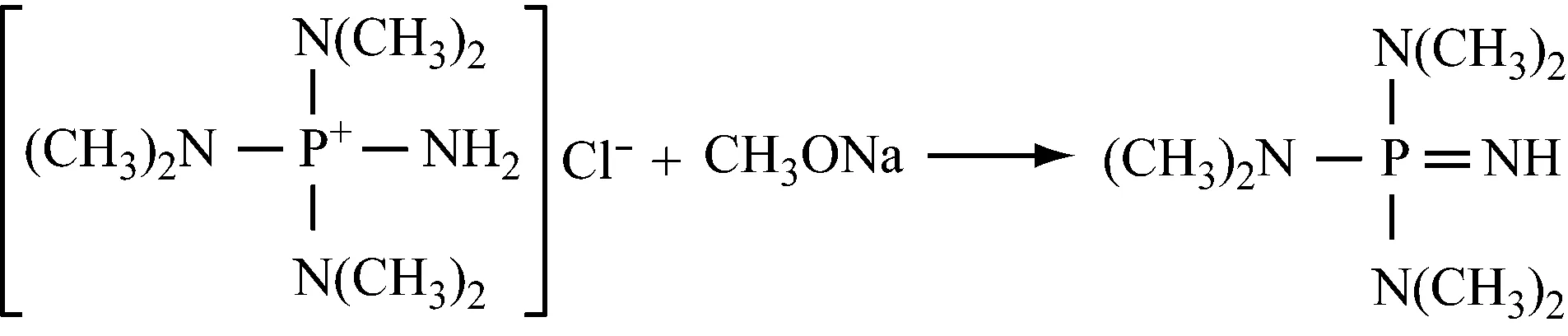
(3) 双(二取代氨基)氯代次磷酸盐的制备
1 mol O=PCl3溶于乙醚中,缓慢滴加4 mol仲胺,在125 ℃下反应24 h,得到双(二取代氨基)氯代次磷酸盐,其结构式为(NR2)2ClP=O。

(4) 磷腈结构的调整
用双(二取代氨基)氯代次磷酸盐与亚氨基正磷反应,再对其进行氯化,然后用NH3处理产物,最后与碱反应,得到(NR2)3P=NP(NR2)2=NH。
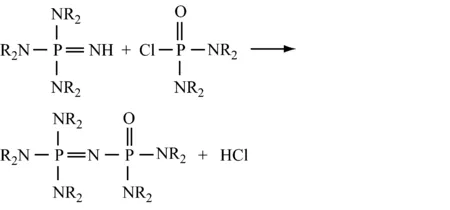
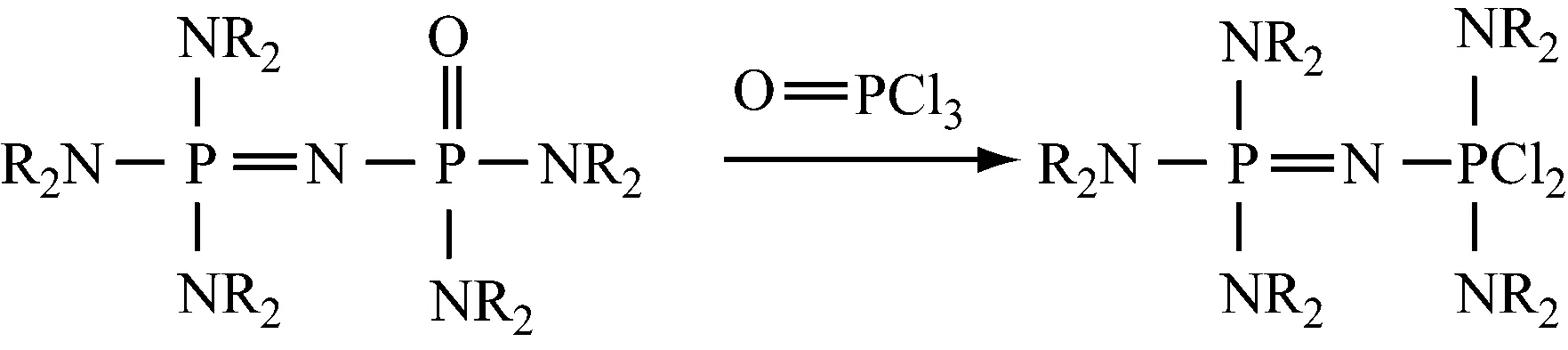
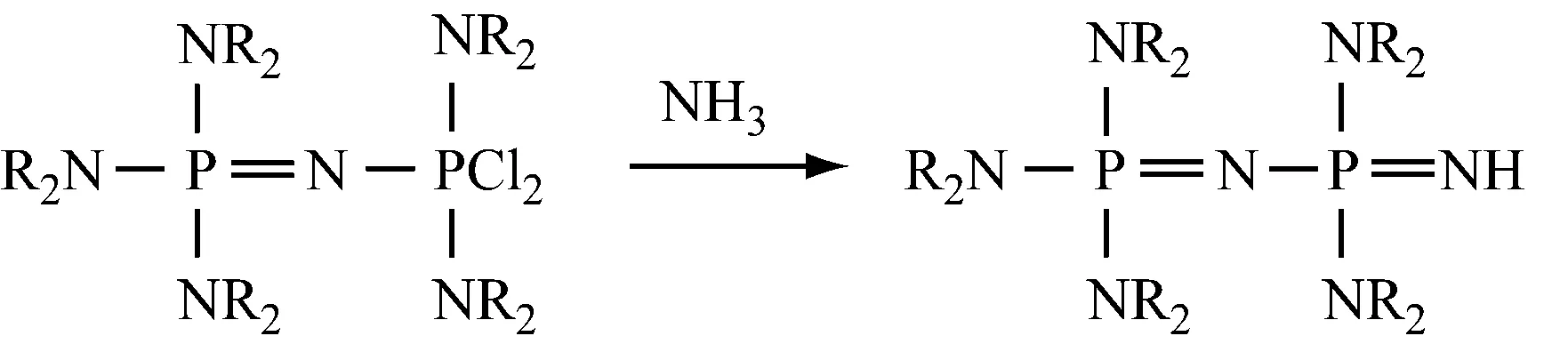
再用该产物代替(NR2)3P=NH,重复上述操作,得到通式为NR2[P(NR2)2=N]aH的化合物,式中:a=0~3,如下所示:
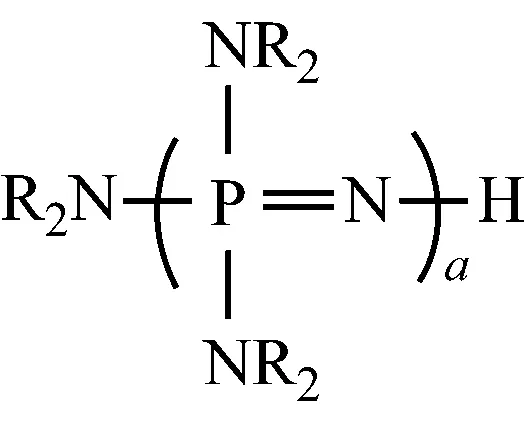
(5) 叔丁基三氯化磷腈的制备
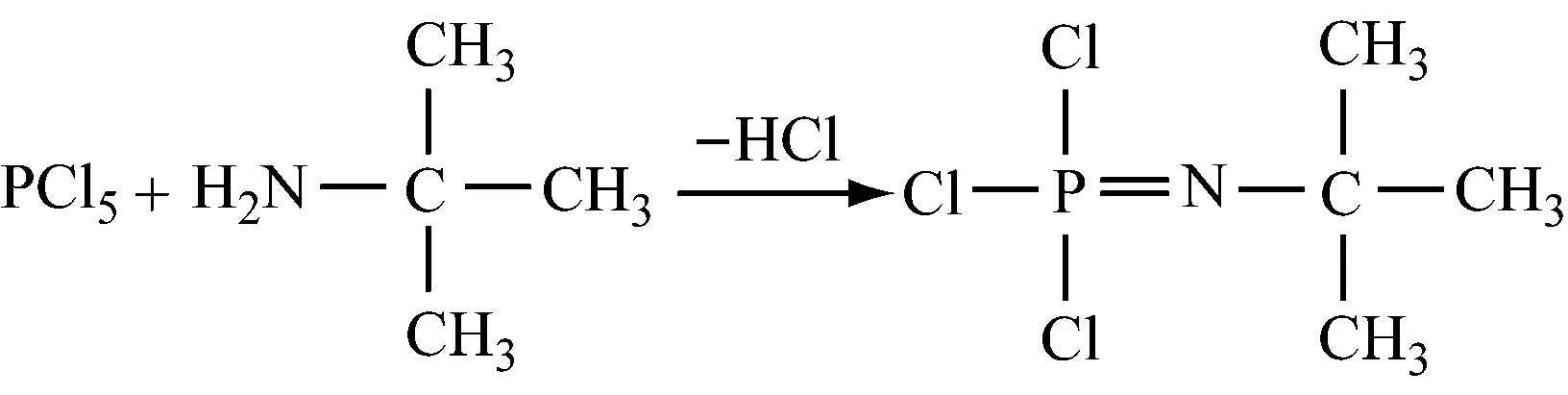
(6) 磷腈化合物的制备
在有机溶剂中,1 mol PCl3=NC(CH3)3与6 mol NR2[P(NR2)2=N]aH反应,得到磷腈化合物。
4.3磷腈盐催化剂的制备[37]
原料:NH3、NaOH、PCl3、(NR2)2P=OCl、HNR2、O=PCl3。
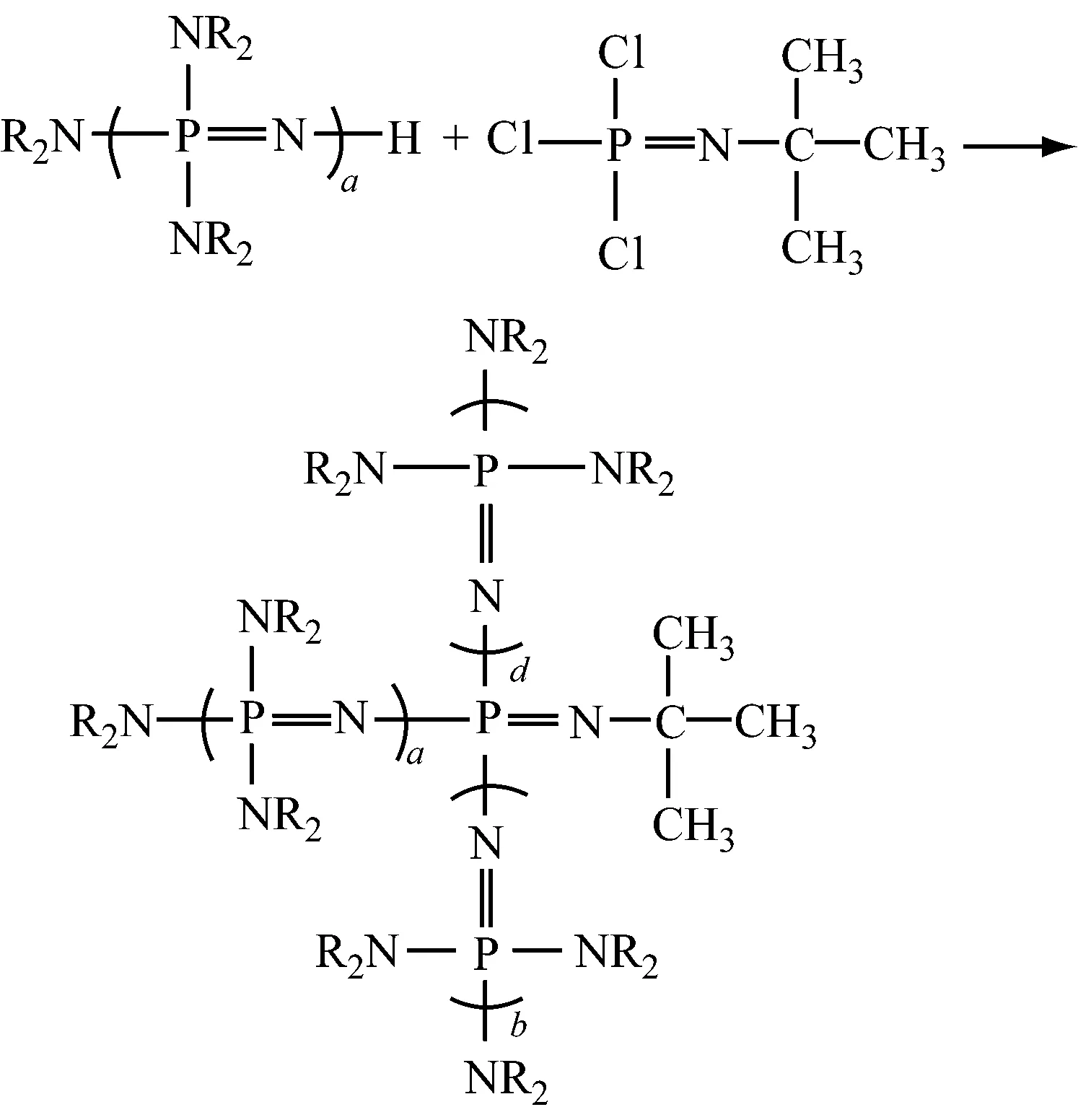
(磷腈化合物)
1 mol PCl5与4 mol不同R和a的磷腈化合物在烃类溶剂中反应,得到无机磷腈盐,阴离子为Cl-。在烃、磷等有机溶剂中,无机磷腈盐与活泼氢化合物的碱金属盐的醇溶液(醇钠/醇溶液)反应制得有机磷腈盐。
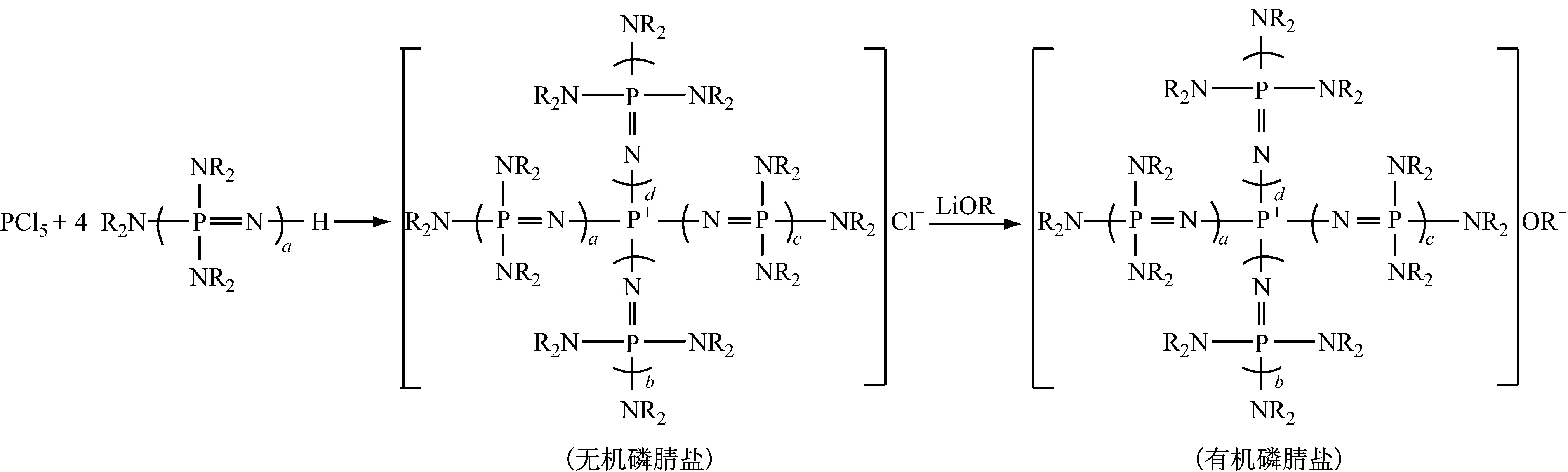
在上述反应过程中, (NR2)3P=NH能与副产物HCl反应生成[(NR2)3PNH2]+Cl-。由于后者不溶于有机溶剂,过滤分离后,在水或极性有机溶剂中用碱处理该盐,能再生为(NR2)3P=NH,重复使用。
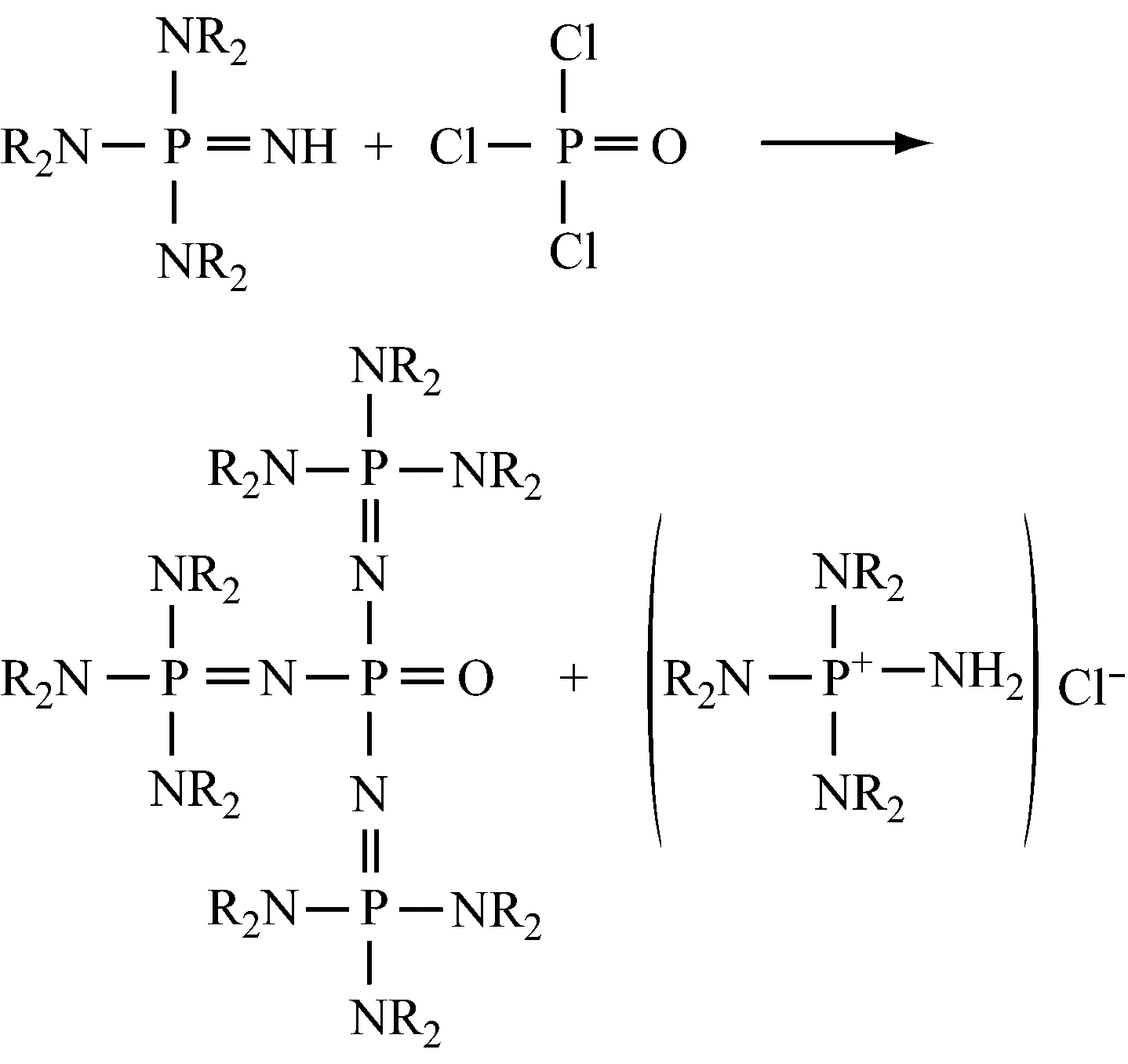
4.4氧化磷腈的制备
原料:NH3、NaOH、HNR2、O=PCl3、PCl5(沸点160 °C)。
首先,在20 ℃,30 min左右,将6 mol的三氯氧磷滴加到1 mol的亚氨基正磷的有机溶液中,并同时搅拌混合物;然后分离出副产物氨基-三(二甲氨基)氯化磷沉淀;再用有机溶剂洗涤沉淀,浓缩滤液,最后用少量有机溶剂将残余物重结晶。
三井化学公司Noborit T等[38]使用非质子型硝基苯溶剂合成氧化磷腈(一种在空气中极易吸水形成水合物的磷腈衍生物)。待反应完成,过滤分离出沉淀,然后于15~40 ℃下用水洗涤,静置分液,脱除水相,减压去除溶剂,得到目的产物(固体),收率达到90%左右。采用这种方法,氧化磷腈几乎不进入水相,也可用重结晶法提纯。
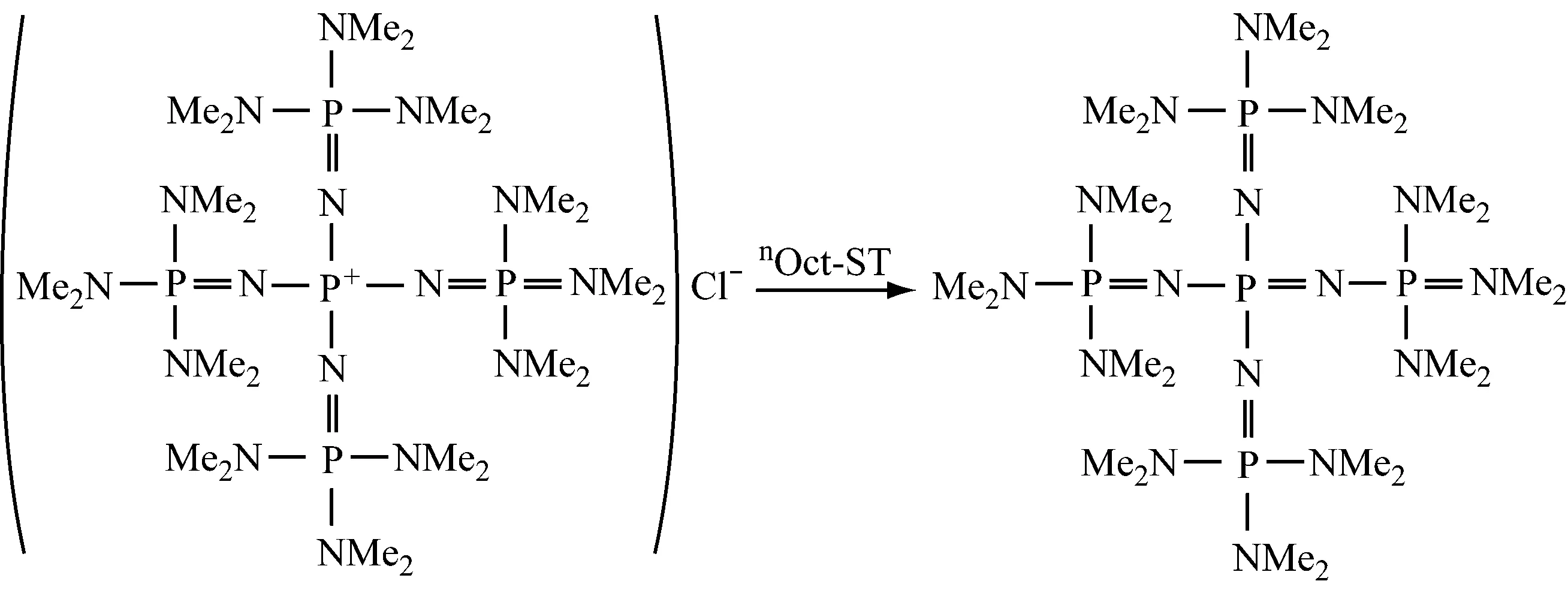
4.5催化剂或催化方法的改进
(1) 负载
在有机反应中,负载催化剂是极其有效的。由于回收再使用不会降低活性,经济效益明显。
Yoshimuran N等[39]在专利中介绍了磷腈负载催化剂,其机制如下所示:
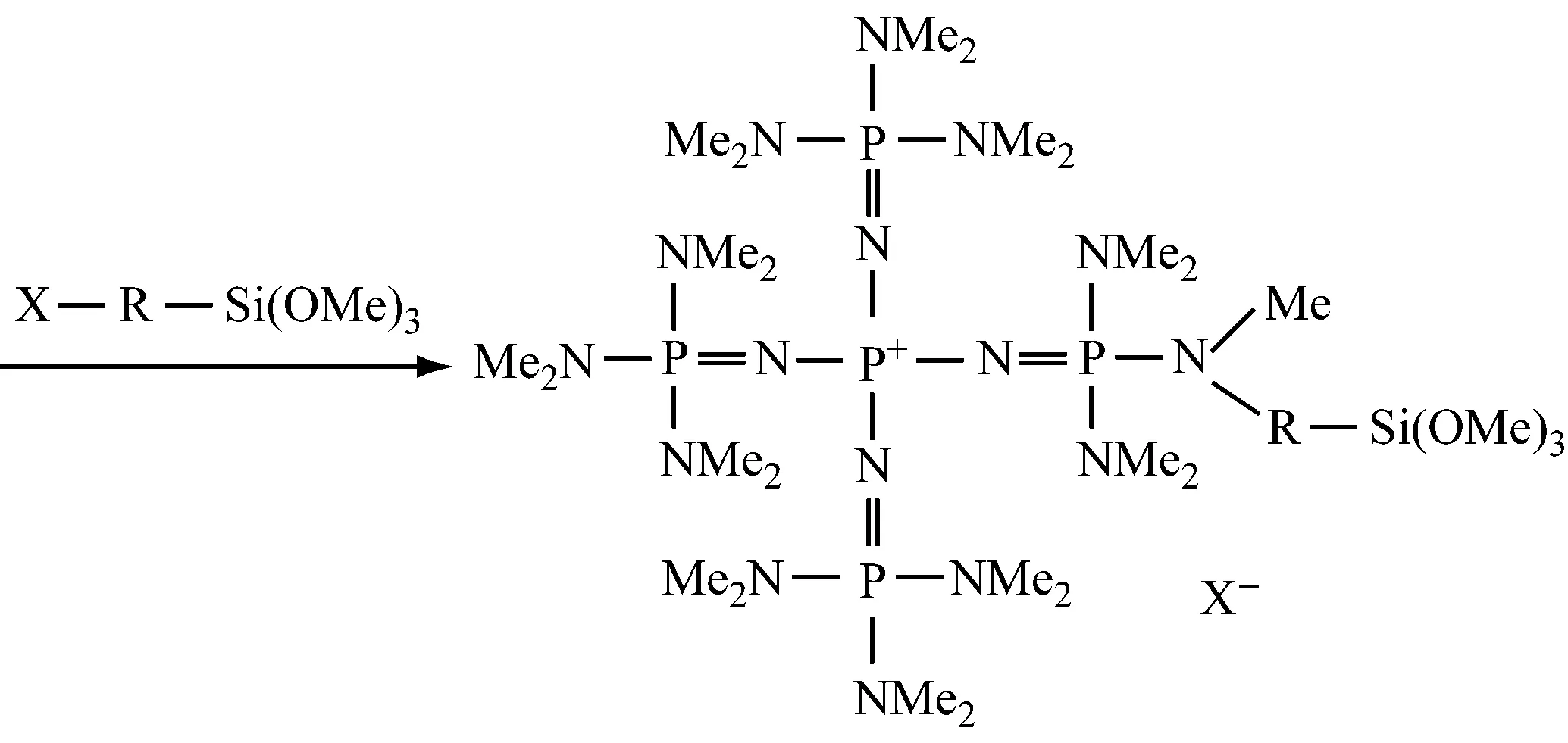
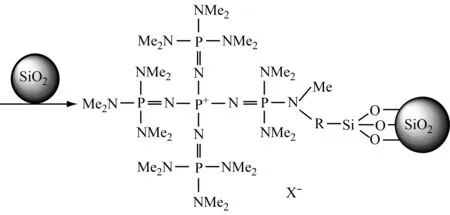
(2) 活化单体和引发剂/链端的协同催化体系
目前催化剂大都是单一体系的催化剂:一类是作为亲电试剂活化单体的催化剂[40],如阳离子催化体系、吡啶、膦以及氮杂环卡宾等;另一类是通过活化引发剂/链端的催化剂,如磷腈等。
需要开发两类催化剂:① 单一催化剂。该催化剂既可以活化引发剂/链端,也可以激活单体,即一部分通过氢键作用活化引发剂/链端,而另一部分作为亲电试剂活化单体,双重活化下达到开环目的。② 复合催化剂,即两种催化剂同时发生作用,一种可以活化引发剂/链端;另一种可以激活单体,两者可以起协同作用,而又不相互反应。
5 结语
与阴离子聚合、阳离子聚合和双金属氰化物聚合等传统催化剂相比,新型的磷腈催化剂更具优势,它可以通过活化引发剂来引发环氧化物开环聚合,克服了双金属氰化物催化剂不能使用小分子作为引发剂的缺点,以及可以使用环氧乙烷封端,提高了聚合效率,简化了工艺流程。另外,磷腈催化剂的负载和活化单体与引发剂/链端的协同催化体系还需要进一步研究。
[1]张荣明, 单红曼, 赵民, 等. 阳离子聚合物PEA的合成与性能[J]. 石油化工高等学校学报, 2012, 25(5): 48-51.
[2]JACK M. Method of making a polyether using a double metal cyanide complex compound: U S, 3 278 457[P]. 1966-10-11.
[3]ROBERT J B. Method of making a polyether using a double metal cyanide complex compound: U S, 3 278 458[P]. 1966-10-11.
[4]黄亦军, 戚国荣, 封麟先. 双金属氰化物络合物催化环氧烷烃开环聚合的特征[J]. 高分子学报, 2002, 1(3): 271-276.
[5]HEROLD R J,LIVIGNI R A. Polymerization kinetics and technology [M ]. Washington D C: ACS, 1973: 208.
[6]KUYPER J, BOXHOORN G. Hexacyanometallate salts used as alkene-oxide polymerization catalysts and molecular sieves[J]. Journal of Catalysis, 1987, 105(1): 163-174.
[7]李学福, 朱东升, 张越涛, 等. Zn-Fe 氰化物配合物的合成及其催化环氧丙烷开环聚合[J]. 吉林大学学报: 理学版, 2006, 44(3): 480-484.
[8]WOJDEL J C, BROMLEY S T, ILLAS F, et al. Development of realistic models for double metal cyanide catalyst active sites[J]. Journal of Molecular Modeling, 2007, 13(6-7): 751-756.
[9]吴立传, 余爱芳, 张敏. 双金属催化环氧化物聚合诱导期的研究[J].高分子材料科学与工程, 2004, 20(5): 84-88.
[10]吴立传, 余爱芳, 张敏.双金属催化环氧化物聚合动力学研究[J].高分子学报, 2003, 2(6): 817-874.
[11]陈华, 朱新宝, 王康芳, 等. 环氧化合物开环聚合催化剂的研究进展[J]. 化学工业与工程技术, 2008, 29(2): 40-45.
[12]胡冰, 黎松, 陈成, 等. 低不饱和度聚醚多元醇伯羟基含量的提高[J]. 聚氨酯工业, 2002, 17(1): 41-44.
[13]于剑昆. 磷腈类催化剂及其在聚醚多元醇合成中的应用[J]. 聚氨酯工业, 2004, 19(5): 10-14.
[14]张治国, 尹红. 环氧乙烷和环氧丙烷开环聚合[J]. 化学进展, 2007, 19(1): 145-152.
[15]张治国,尹红.环氧乙烷环氧丙烷开环聚合反应动力学研究[J].化学进展,2007,19(4):575-582.
[16]SIMONS D M, VERBANC J J. The polymerization of propylene oxide[J]. Journal of Polymer Science, 1960, 44: 303-311.
[17]GEE G, HIGGINSON W C E, TAYLOR K J, et al. The polymerization of epoxides(Part III). The polymerization of propylene oxide by sodium alkoxides[J]. Journal of the Chemical Society (Resumed), 1961: 4298-4303.
[18]李小童, 张杰, 尚丙坤, 等. 端烯丙基聚醚制备中醇盐化试剂的选择及产物的精制研究[J]. 化学推进剂与高分子材料, 2005, 3(3): 21-23.
[19]王志新. DMC催化剂及其在聚醚多元醇生产中的应用[J]. 聚氨酯工业, 2002, 17(3): 41-43.
[20]王西奎, 刘汉霞, 顾尧, 等. 双金属氰化物络合物催化剂的制备, 表征与催化性能研究[J]. 现代化工, 2003(z1): 126-128.
[21]黄亦军. 双金属氰化物络合物催化环氧烷烃开环聚合的研究[D]. 杭州:浙江大学, 2002.
[22]慕朝师, 黄科林, 李克贤, 等. 聚醚多元醇的研究进展[J]. 化工技术与开发, 2009, 38(12): 13-18.
[23]戚渭新, 丁国来, 韩勇, 等. 双金属氰化物络合催化合成聚醚起始条件的研究[J]. 聚氨酯工业, 2003, 18(2): 25-27.
[24]万小龙, 罗钟瑜, 胡忠伟. 聚合物多元醇合成引入整理剂的原位聚合法[J]. 黎明化工, 1990(1): 35.
[25]李茂元. 低不饱和度聚醚多元醇及其催化剂的制备工艺[J]. 聚氨酯工业, 1999, 14(4): 43-46.
[26]NOBORI T, HAYASHI T, SHIBAHARA A, et al. Development of novel molecular catalysts “phosphazene catalysts” for commercial production of highly advanced polypropylene glycols[J]. Catalysis Surveys from Asia, 2010, 14(3-4): 164-167.
[27]李传碧. 金属离子负载修饰阳离子树脂催化合成环己烯活性的研究[J]. 吉林师范大学学报: 自然科学版, 2005, 26(3): 22-23.
[28]李灿. 高度隔离过渡金属催化剂及其催化烯烃环氧化反应[J]. 催化学报, 2001, 22(5): 479-483.
[29]万惠霖, 袁友珠, 高景星, 等. 过渡金属配合物催化剂及其分子设计构思的发展与相互作用[J]. 高等学校化学学报, 1997, 18(7): 1185-1193.
[30]于剑昆. 三井化学公司新型聚醚多元醇的制备与应用[J]. 化学推进剂与高分子材料, 2005, 3(5): 8-13.
[31]陈华, 朱新宝, 房连顺, 等. 磷腈类催化剂研究进展[J]. 江苏化工, 2008(2): 2.
[32]黄茂松, 贾润萍. 中国汽车用聚氨酯材料发展方向[J]. 聚氨酯, 2012(3): 23.
[33]刘佳, 程斌. 环氧乙烷环氧丙烷共聚醚的研究进展[J]. 高分子通报, 2008(10): 13-20.
[34]于剑昆, 陈运铎. 用磷腈类催化剂合成的新型聚醚多元醇及其在聚氨酯泡沫制备中的应用新进展[J]. 化学推进剂与高分子材料, 2012, 10(2): 1-7.
[35]ILLY N, BOILEAU S, PENELLE J, et al. Metal-free activation in the anionic ring-opening polymerization of cyclopropane derivatives[J]. Macromolecular Rapid Communications, 2009, 30(20): 1731-1735.
[36]YANG H, YAN M, PISPAS S, et al. Synthesis of poly [(ethylene carbonate)-co-(ethylene oxide)] copolymer by phosphazene-catalyzed ROP[J]. Macromolecular Chemistry and Physics, 2011, 212(23): 2589-2593.
[37]NOBORI T, SUZUKI T, KIYONO S, et al. Phosphazenium salt and preparation process thereof, and process for producing poly(alkylene oxide): EP, 791600[P]. 1997-08-27.
[38]NOBORI T, KOUNO M, SUZUKI T, et al. A polymerization catalyst for forming a polyether: U S, 5 990 352[P]. 1999-11-23.
[39]YOSHIMURA N, KIYONO S, MIZUTANI K, et al. Novel phosphazene-supported catalyst, novel compound thereof and use thereof: U S, Patent Application 13/198 397[P]. 2011-08-04.
[40]许茸. 含磷类双功能有机小分子催化ε-己内酯聚合反应的研究[D]. 哈尔滨:东北林业大学, 2012.
Research Progress on Catalysts for Synthesis of Polyether Polyol
ZHAO Rui-chao, YAO Li-na, GAI Dong-jie, GU Yao
(Key Laboratory of Ministry of Education of Rubber Materials & Engineering,Qingdao University of Science & Technology, Qingdao 266042, China)
The mechanism of the anion polymerization, cation polymerization, double metal cyanide complex catalyzed polymerization and new phosphazene polymerization in catalyzing epoxide is reviewed, and the preparation and development trend of phosphazene catalysts are introduced in detail.
cation polymerization; anionic polymerization; double metal cyanide complex catalyst; phosphazene catalyst
赵瑞超(1990-),男,硕士,从事功能及特种高分子材料的研究。
TQ 320
A
1009-5993(2016)01-0016-10
2015-10-27)
猜你喜欢
氯碱工业(2022年5期)2022-11-25 15:44:27
中国氯碱(2021年10期)2021-04-13 15:56:36
世界农药(2019年2期)2019-07-13 05:55:12
电子测试(2018年1期)2018-04-18 11:52:24
化工生产与技术(2016年5期)2016-03-13 10:07:26
合成化学(2015年4期)2016-01-17 09:01:04
海军航空大学学报(2015年1期)2015-11-11 17:22:41
股市动态分析(2015年12期)2015-09-10 13:18:31
石油化工技术与经济(2014年5期)2014-04-06 01:05:12
火炸药学报(2014年1期)2014-03-20 13:17:24
