
QuEChERS方法净化UPLC-MS检测猪肉中35种兽药残留
2016-10-18 06:25梁飞燕张科明卢日刚
安徽农业科学 2016年25期
梁飞燕,张科明,卢日刚
(1.广西-东盟食品药品安全检验检测中心,广西南宁 530021;2.广西食品药品检验所,广西南宁 530021)
QuEChERS方法净化UPLC-MS检测猪肉中35种兽药残留
梁飞燕1,张科明2,卢日刚2
(1.广西-东盟食品药品安全检验检测中心,广西南宁 530021;2.广西食品药品检验所,广西南宁 530021)
[目的]建立QuEChERS方法提取和净化、超高效液相色谱串联质谱法(UPLC-MS)检测猪肉中35种兽药残留的方法。[方法]猪肉样品分别用Na2EDTA-Mcllvaine(pH 4.0)溶液和2.5%乙酸乙腈提取后,得到的溶液以NaCl和无水Na2SO4为脱水剂,-NH2为吸附剂进行净化浓缩后,在电喷雾正离子模式下,以多反应监测(MRM)方法进行检测,基质空白加标曲线外标法进行定性和定量分析。[结果]35种兽药在0.5~50.0 ng/mL浓度范围内线性关系良好,相关系数均大于0.995,4个不同浓度添加水平的平均回收率为60%~110%,相对偏差(RSD)均小于10%,检出限(LOD,S/N=3∶1)为0.02~1.20 μg/kg,定量限(LOQ,S/N=10∶1)为0.07~4.00 μg/kg。[结论]该方法简单、快速、可靠、灵敏、高效,可用于猪肉中多兽药残留的分析检测和监控。
兽药残留;检测;QuEChERS;超高效液相色谱-串联质谱法
兽药残留(residues of veterinary drug)是“兽药在动物源食品中的残留”的简称,根据联合国粮农组织和世界卫生组织(FAO/WHO)食品中兽药残留联合立法委员会的定义,兽药残留是指动物产品的任何可食部分所含兽药的母体化合物及(或)其代谢物,以及与兽药有关的杂质。滥用兽药极易造成动物源食品中有害物质的残留,这不仅对人体健康造成直接危害,而且对畜牧业的发展和生态环境也造成极大危害。动物源食品中的兽药残留已逐渐成为全世界关注的一个焦点。因此,严格监控动物源性食品中的兽药残留具有重要的意义。
目前,我国关于动物源性食品中兽药残留检测的国家标准[1-4]大部分都是以分类检测为主,一个样本要进行多次检验,需要耗费极大的人力、物力、财力和时间。同时,动物源性食品种类繁多,样品基质复杂,检测目标物种类和组分多,理化性质差异大,是兽药残留分析检测技术的重大难题。近年来,关于兽药残留检测报道的相关文献中,样品前处理技术主要有:固-液萃取法[5-8]、固相萃取法[9-10]、基质固相分散技术[11-12]、QuEChERS方法[13-14]等,检测技术有:高效液相色谱法[15-16]、液相色谱-串联质谱法[17-18]、液相色谱-四级杆-飞行时间质谱法[19-20]。QuEChERS方法是近年来发展起来的样品前处理技术,具有快速、简易、环保、价廉的优点,而液相色谱-串联质谱法同时具备了色谱和质谱的优点,对基质复杂的样品具有选择性强、灵敏度高、分离能力强以及能提供分子量和结构信息的优点,已成为当前检测兽药残留的重要手段。常用的兽药有磺胺类及其增效剂、β-受体激动剂、四环素类、喹诺酮类、抗病毒类、孕激素类、蛋白同化激素类等,此类兽药极性差异大,难以用一种方法同时进行检测分析,现鲜有文献报道此类兽药残留同时测定的方法。因此,为简化分析检测方法,提高提取和净化效率,提高检测灵敏度,建立快速、有效、高通量的多兽药残留分析方法,笔者对猪肉中的多兽药残留进行了考察,建立了QuEChERS方法净化,UPLC-MS方法同时检测了磺胺类及其增效剂、β-受体激动剂、四环素类、喹诺酮类、抗病毒类、孕激素类、蛋白同化激素类共7类35种兽药。
1 材料与方法
1.1材料
1.1.1标准品。19种磺胺类及其增效剂:磺胺甲噻二唑、磺胺二甲异噁唑、磺胺氯哒嗪、磺胺噻唑、磺胺间甲氧嘧啶、磺胺甲基嘧啶、磺胺邻二甲氧嘧啶、磺胺吡啶、磺胺对甲氧嘧啶、磺胺二甲嘧啶、磺胺间二甲氧嘧啶、磺胺二甲异嘧啶钠盐、琥珀酰磺胺噻唑、磺胺喹噁啉、磺胺噁唑,购自Dr.Ehrenstorfer GmbH;磺胺嘧啶、磺胺甲(基异)噁唑、磺胺苯吡唑、甲氧苄啶,购自中国食品药品检定研究院。5种β-受体激动剂:盐酸克伦特罗、莱克多巴胺(盐酸盐)、沙丁胺醇、克仑潘特盐酸盐,购自Dr.Ehrenstorfer GmbH;马喷特罗盐酸盐,购自WITEGA。4种四环素类:四环素盐酸盐、土霉素、金霉素盐酸盐,购自Dr.Ehrenstorfer GmbH;美满霉素,购自中国食品药品检定研究院。3种喹诺酮类:诺氟沙星、氧氟沙星、恩诺沙星,购自中国食品药品检定研究院。1种抗病毒类:金刚烷胺,购自中国食品药品检定研究院。1种孕激素类:黄体酮,购自中国食品药品检定研究院。2种蛋白同化激素类:睾酮、甲睾酮,购自中国食品药品检定研究院。
1.1.2主要试剂与耗材。柠檬酸、磷酸氢二钠、磷酸、冰醋酸、氯化钠、无水硫酸钠,均为分析纯,国药集团化学试剂有限公司;乙二胺四乙酸二钠,分析纯,广东光华科技股份有限公司;-NH2吸附剂,实验室专用,安谱;乙腈、甲酸,色谱纯,德国默克公司;试验用水,GB/T 6682规定的一级水;去离子水,美国密理博公司;微孔滤膜(0.22 μm、尼龙),安谱;15、50 mL塑料离心管,CNW。
1.1.3主要仪器设备。安捷伦1290超高效液相色谱仪串联Triple Quad 5500三重四级杆串联质谱仪;FS25匀浆机,福禄克公司;VORTEX GENIE-2涡旋振荡仪,SCIENTIFIC INDUSTRIES,INC.;S180H超声波清洗器,艾尔玛公司;ROTANTA 460R离心机,海蒂诗公司;N-EVAPTM112氮吹仪,美国Organomation Associates,Inc.。
1.1.4溶液的配制。Mcllvaine缓冲溶液:取0.1 mol/L柠檬酸溶液1 000 mL与0.1 mol/L Na2HPO4溶液625 mL混匀。Na2EDTA-Mcllvaine缓冲溶液(pH 4.0):称取60.5 g乙二胺四乙酸二钠放入1 625 mL Mcllvaine缓冲溶液中,振摇使其溶解,用磷酸调节pH为4.0。
1.2方法
1.2.1样品前处理。称取匀浆后的猪肉样品2.0 g于50 mL离心管中,加入Na2EDTA-Mcllvaine缓冲液(pH 4.0)5.0 mL,摇匀,超声5 min,涡旋1 min,10 000 r/min离心5 min,取上清液置另一50 mL离心管中,残渣再精密加入2.5%乙酸乙腈溶液20 mL,摇匀,超声5 min,涡旋1 min,10 000 r/min离心5 min,合并2次提取的上清液置同一50 mL离心管中,加入NaCl 2.0 g,无水Na2SO42.0 g,涡旋5 min,5 ℃10 000 r/min离心5 min。精密吸取上清液10 mL置15 mL离心管中,加入-NH2吸附剂200 mg,涡旋5 min,5 ℃ 10 000 r/min离心5 min。精密吸取上清液5.0 mL置15 mL离心管中,50 ℃氮气吹干,加入0.1%甲酸-25%乙腈水溶液1.0 mL,超声5 min,涡旋1 min,用0.22 μm滤膜过滤,取滤液上机测定。
1.2.2色谱-质谱条件。色谱条件:XBridge® BEH C18,2.5 μm,4.6×100 mm;流动相为A,0.1%甲酸水溶液;B,0.1%甲酸乙腈溶液;梯度洗脱程序:0~2.0 min,95%~80% A;2.0~4.0 min,80%~75% A;4.0~7.0 min,75%~45% A;7.0~11.0 min,45%~10% A;11.0~16.0 min,10% A;流速0.5 mL/min;柱温为40 ℃;进样量5 μL。
质谱条件:扫描模式为正离子(ESI+);采集方式为多反应监测(MRM);离子源温度650 ℃,雾化气压力(GSI)3.79×105Pa;辅助气压力3.79×105Pa;气帘气压力(CUR):2.41×105Pa;碰撞气体速度为中流速;电喷雾电压(IS)为5 500 V;去簇电压(DP)130.0 V、碰撞室入口电压(EP)为10.0 V、出口电压(CXP)13.0 V;35种目标化合物母离子、碎片离子(定量离子、定性离子)及其碰撞电压(CE)详见表1。
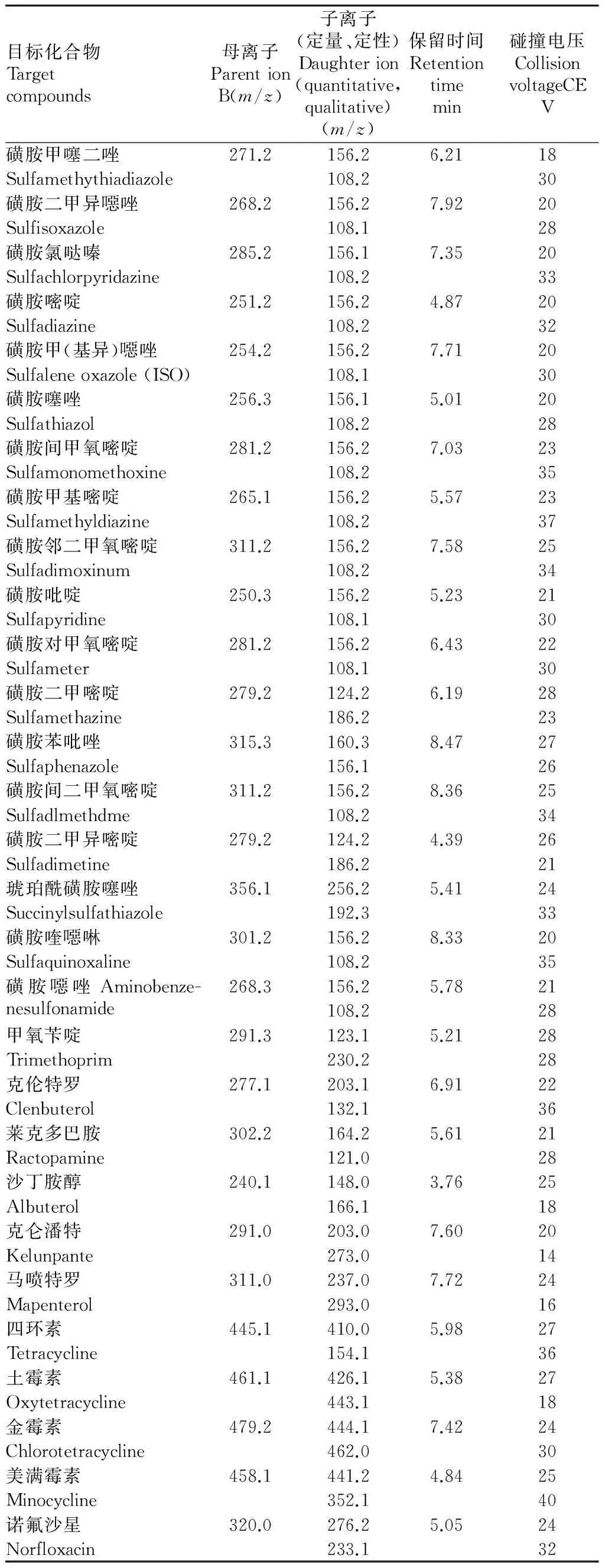
表1 35种目标化合物质谱参数
接下表
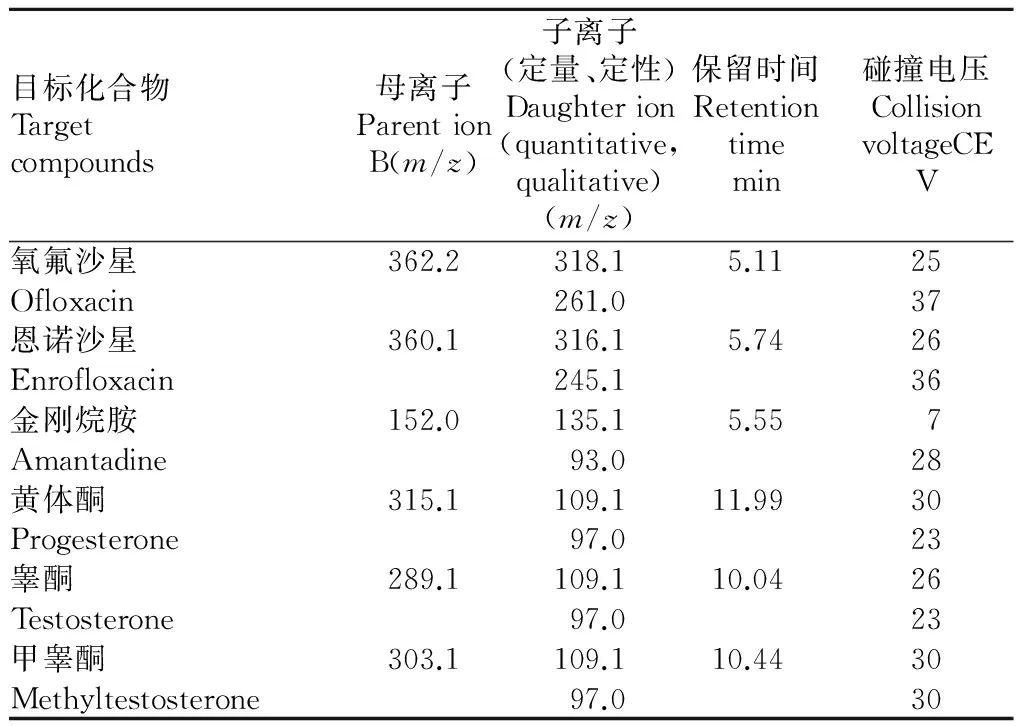
续表1
2 结果与分析
2.1仪器条件的优化
2.1.1流动相的优化。流动相的离子强度和酸碱度均会影响目标物的峰形和分离效果。通过查阅相关文献[21-23],该试验考察了6种流动相:①0.01 mol/L乙酸铵溶液(用甲酸调节pH至4.5)-乙腈;②0.01 mol/L乙酸铵溶液(用甲酸调节pH至4.5)-甲醇;③0.01 mol/L乙酸铵溶液(用甲酸调节pH至4.5)-0.01 mol/L乙酸铵乙腈溶液;④0.1%甲酸溶液-乙腈;⑤0.1%甲酸溶液-甲醇;⑥0.1%甲酸溶液-0.1%甲酸乙腈溶液。结果发现:当流动相为0.1%甲酸溶液-0.1%甲酸乙腈溶液时(图1),各化合物的峰形对称性好,灵敏度高,基线噪音明显降低,故选择了0.1%甲酸溶液-0.1%甲酸乙腈溶液作为流动相。
图1 流动相⑥分离效果Fig.1 The isolation effect of mobile phases ⑥
2.1.2色谱柱条件的优化。该试验考察了如下色谱柱:CORTECSTMC18,2.7 μm,4.6×100 mm;XBridge® BEH C18,2.5 μm,4.6×100 mm;XBridgeTMphenyl,3.5 μm,2.1×150 mm;ACQUITY UPLC® CSHTM C18,1.7 μm,3.0×150 mm;Poroshell 120 EC-C18,2.7 μm,4.6×50 mm;CAPCELL PAK C18MGⅢ-H,3 μm,2.0 mm I.D.×100 mm;Inertsil C8-3,2 μm,2.1×100 mm。通过比较发现,C8和phenyl填料对目标物的保留能力弱,峰形较宽,分离度不好;CORTECS、MGⅢ对大部分磺胺类、喹诺酮类的保留性均较差;CSH、Poroshell虽然对目标物的保留能力好,但是峰形较宽不理想;采用BEH的C18色谱柱时,35种目标物的保留能力好,峰形对称性良好,故该试验采用了BEH的C18色谱柱。
2.2样品前处理条件的优化
2.2.1提取溶剂的选择。该试验查阅了相关文献[24-25],考察了4种不同的溶剂进行提取:①0.1 mol/L柠檬酸-0.2 mol/L Na2HPO4溶液(8∶5,V∶V,pH 4);②Na2EDTA-Mcllvaine缓冲溶液(pH 4.0);③5%的乙酸乙腈;④5%的氨水乙腈。结果发现,当提取溶剂中含有络合剂时,由于络合了与四环素类和喹诺酮类化合物中螯合的金属离子,使得四环素类和喹诺酮类被释放出来,提取回收率明显提高;磺胺类、四环素类和喹诺酮类化合物在碱性条件时会降解,提取回收率大大降低;提取溶剂中适当的酸度,有利于抗病毒类、孕激素、蛋白同化激素类化合物的提取;β-受体激动剂、抗病毒类、孕激素、蛋白同化激素在酸、碱条件下均比较稳定,提取效果相当。
鉴于35种化合物理化性质差异大,用一种溶剂很难将全部化合物提取完全,故该试验还考察了2种溶剂复提取的方式:①0.1 mol/L柠檬酸-0.2 mol/L Na2HPO4溶液(8∶5,V∶V,pH 4)→5%的乙酸乙腈;②Na2EDTA-Mcllvaine缓冲溶液(pH 4.0)→5%的乙酸乙腈。通过试验发现,2种溶剂复提取的提取效果均优于单一溶剂提取的效果,采用含有络合剂的Na2EDTA-Mcllvaine缓冲溶液(pH 4.0)→5%的乙酸乙腈作为提取溶剂,35种化合物的提取回收率比较理想。各种提取溶剂的提取回收率如图2所示。
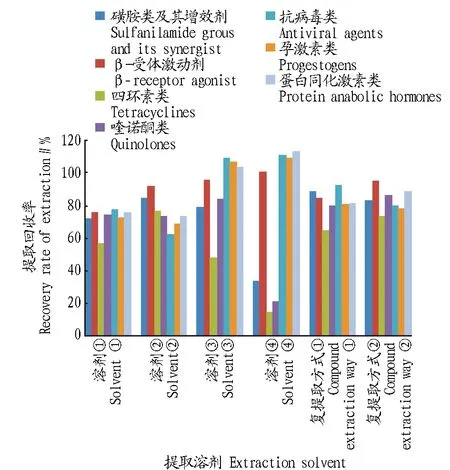
图2 6种提取溶剂的平均提取回收率Fig.2 The average recovery of 6 extraction solvents
由于β-受体激动剂、四环素类、喹诺酮类、抗病毒类、孕激素类、蛋白同化激素类中含有碱性基团,适当的酸度更利于上述化合物的提取。为此该试验还考察了1.0%、2.5%、5.0%、7.0%的乙酸乙腈的提取效果,发现磺胺类和四环素类化合物对酸度比较敏感,酸度过低,电离程度低而使得提取回收率偏低;当酸度>5%时,部分磺胺可能会降解,故该试验采用了2.5%的乙酸乙腈。
2.2.2净化条件的优化。QuEChERS方法意为:快速(quick)、简单(easy)、便宜(cheap)、高效(effective)、耐用(rugged)和安全(safe)[26],该法常用的吸附剂有:C18、-NH2吸附剂、中性氧化铝、酸性氧化铝、硅藻土、硅镁吸附剂、乙二胺-N-丙基丙烷(PSA)/硫酸镁。通过对上述吸附剂的净化效果和吸附作用进行考察,结果如图3所示:中性氧化铝、酸性氧化铝、乙二胺-N-丙基丙烷(PSA)/硫酸镁、硅镁吸附剂、硅藻土对四环素类、喹诺酮类化合物、孕激素均有不同程度的吸附,且净化效果不理想,基线噪音大,干扰峰多。C18和-NH2吸附剂对35种目标化合物的吸附能力相对较小,提取回收率良好,但-NH2吸附剂净化效果比C18的明显,杂质峰干扰少。同时该试验还考察了吸附剂用量对净化效果和提取回收率的影响:100 mg-NH2吸附剂、200 mg-NH2吸附剂、400 mg-NH2吸附剂、200 mg-NH2吸附剂+200 mg C18,结果发现,吸附剂用量对于响应小、灵敏度低的物质如四环素类影响比较大,当用量为200 mg-NH2吸附剂时,基线噪音小,净化效果明显,提取回收率良好,故该试验采用了200 mg-NH2做为吸附剂。
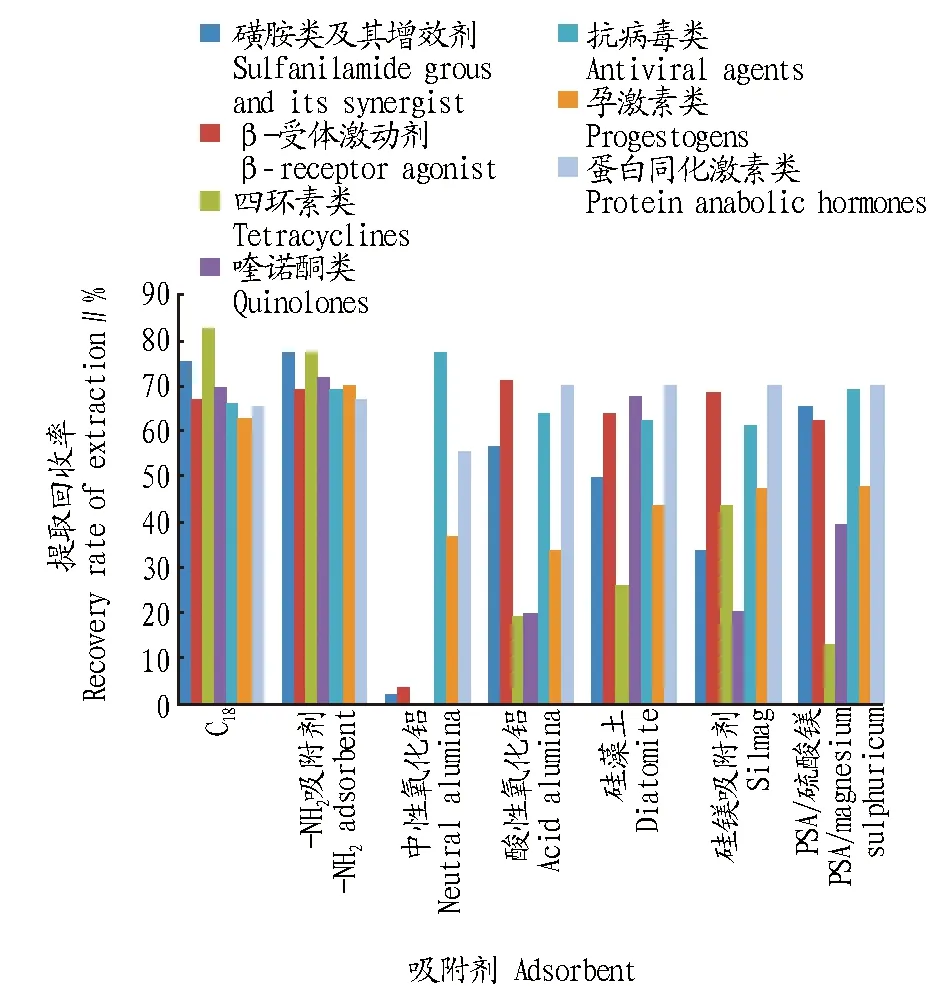
图3 7种吸附剂的平均提取回收率Fig.3 The average recovery of 7 adsorbents
2.2.3复溶溶剂的选择。复溶溶剂中有机相的比例和酸碱度均会影响目标化合物溶解,同时进样时还会影响到色谱的基线和峰形,即造成溶剂效应。该试验根据各目标化合物的溶解性能,考察了以下溶剂:①0.1%甲酸溶液-0.1%甲酸乙腈(85∶15),②水-乙腈(85∶15),③0.1%甲酸-25%乙腈水溶液,④0.1%甲酸-50%乙腈水溶液。通过比较发现,甲酸的存在,更利于含有碱性基团物质的溶解;当有机相中乙腈的浓度大于25%时,非极性物质如孕激素、蛋白同化类激素能更好地溶解,故该试验采用了0.1%甲酸-25%乙腈水溶液作为复溶溶剂。
2.3基质效应的消除在液相色谱-串联质谱法中,基质效应的产生可能是基质与待分析组分在离子化过程中发生竞争,使得目标化合物的碎片离子生成的效率和离子强度显著地减低或增高,从而影响测量结果的精密度和准确性。为了抵消基质效应对测定结果的影响,该试验对以下方法进行了比较:①纯标准物质线性;②基质空白加标线性:将空白样品按前处理方法提取净化后,在提取液中添加目标化合物配制成系列标准曲线,再按已定的色谱质谱条件进行检测[27];③随行加标线性:取空白样品,加入一定量的标准溶液,与样品同时进行提取和净化得到的线性。用上述3种线性分别对同一加标回收率溶液计算回收率进行比较发现,通过基质空白加标线性的方法能够很好地消除基质带来的影响,得到的线性、加标回收率均能满足食品残留检测的要求。
2.4方法线性、精密度、检出限、定量限和准确度为了消除基质效应,该试验采用了基质空白溶液配制标准曲线。以浓度为横坐标,各化合物提取得到的定量离子峰的峰面积为纵坐标绘制标准曲线,计算相关系数,得到35种目标化合物在浓度为0.5~50.0 ng/mL范围内,色谱峰面积和浓度呈良好线性关系,相关系数均在0.995以上;同一标准曲线溶液连续进样6次,各化合物定量离子峰的峰面积RSD均<4%,仪器精密度良好。
同时,取空白猪肉样品,采用逐级稀释的方法添加适量的标准品溶液,按照“1.2.1”项下的步骤同法处理,“1.2.2”项下的色谱-质谱条件进样,得到空白样品加标色谱图,以各化合物定量离子质谱图峰高按照信噪比(S/N)为1∶3、1∶10分别计算检出限(LOD)和定量限(LOQ)。得到35种目标化合物检出限范围为0.02~1.20 μg/kg,定量限范围为0.07~4.00 μg/kg。通过与动物源性食品兽药残留检测方法相关国家标准[1-4]比较,该方法检出限低、灵敏度较高,可满足猪肉中兽药残留检测的要求。
取空白猪肉样品,按照2、4、20、100 μg/kg的浓度水平添加标准品溶液,一式4份,涡旋1 min,静置30 min后按照“1.2.1”项下的步骤同法处理,“1.2.2”项下的色谱-质谱条件进样,根据各组分的峰面积计算加标回收率。同时制备同一添加水平(20 μg/kg)6份用于测定重复性。结果35种目标化合物4个不同浓度添加水平平均回收率在60%~110%,相对标准偏差均小于10%,整体回收率水平和方法重复性良好,符合食品检测的要求。
2.5样品测定对分别购自超市、市场等的10份猪肉样品,采用该试验建立的方法进行测定,同时做加标样品(20 μg/kg)进行质量控制,结果均未发现阳性样品,样品测定色谱图见图4,加标样品提取回收率良好,各目标化合物的提取回收率在62%~83%,加标样品色谱图见图5。
图4 猪肉样品色谱Fig.4 Chromatogram of pork samples
图5 加标样品色谱Fig.5 Chromatogram of spiked sample
3 结论与讨论
该试验建立了QuEChERS净化、结合UPLC-MS方法同时检测猪肉中磺胺类及其增效剂、β-受体激动剂、四环素类、喹诺酮类、抗病毒类、孕激素类、蛋白同化激素类共7类35种兽药的方法。与国家标准相比,该方法克服了单一种类兽药残留检验操作繁琐、检验周期长、成本高的缺点,实现了多类理化性质差异大的兽药残留的同时测定。该方法操作简单、快速、灵敏、准确、高通量,适用于猪肉中多兽药残留的筛查和监测。
[1]杨奕,国伟,吴永宁,等.动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱 质谱法:GB/T 21312—2007[S].北京:中国标准出版社,2008.
[2]邵兵,李晓娟,吴永宁,等.动物源性食品中β-受体激动剂残留检测方法 液相色谱-质谱 质谱法GB/T 21313—2007[S].北京:中国标准出版社,2008.
[3]林黎明,张鸿伟,王凤美,等.动物源性食品中磺胺类药物残留量的测定 高效液相色谱-质谱质谱法:GB/T 21316—2007[S].北京:中国标准出版社,2008.
[4]岳振峰,谢丽琪,叶卫翔,等.动物源性食品中四环素类兽药残留量检测方法 液相色谱-质谱 质谱法与高效液相色谱法:GB/T 21317—2007[S].北京:中国标准出版社,2008.
[5]高玲,张丹,曹军,等.高效液相色谱-串联质谱法测定水产品中大环内酯类药物残留[J].中国兽药杂志,2012,46(4):29-33.
[6]ZHAN J,ZHONG Y Y,YU X J,et al.Multi-class method for determination of veterinary drug residues and othercontaminants in infant formula by ultra performance liquid chromatography-tandem mass spectrometry[J].Food chemistry,2013,138:827-834.
[7]熊春兰,郭平,占春瑞,等.高效液相色谱-串联质谱法同时测定水产品中8类38种兽药残留[J].分析测试学报,2013,32(2):193-198.
[8]DASENAKI M E,NIKOLAOS S.Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder,butter,fish tissue and eggs using liquid chromatography-tandem mass spectrometry[J].Analytica chimica acta,2015,880:103-121.
[9]李晓丽,李娜,李鹏,等.高分子印记固相萃取-液相色谱质谱法测定水产品中孔雀石绿、结晶紫、亮绿及其代谢产物[J].中国食品卫生杂志,2012,24(3):209-214.
[10]曹慧,陈小珍,朱岩,等.多壁碳纳米管固相萃取技术同时测定蜂蜜中多类兽药残留[J].高等学校化学学报,2013,34(12):2710-2715.
[11]王炼,黎源倩,王海波,等.基质固相分散-超高效液相色谱-串联质谱法同时测定畜禽肉和牛奶中20种兽药残留[J].分析化学,2011,39(2):203-207.
[12]秦峰.基质固相分散技术在兽药残留分析中的应用[J].化学通报,2009(2):130-132.
[13]ZHANG Y Q,LI X,LIU X M,et al.Multi-class,multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry[J].Journal of dairy science,2015,98:8433-8444.
[14]宓捷波,许迪明,李淑静,等.奶粉中残留兽药的QuEChERS方法应用研究[J].食品研究与开发,2015,36(2):121-125.
[15]赵亚华,李勇,何学芳.高效液相色谱法同时测定动物源食品中22种兽药残留技术研究[J].中国卫生检验杂志,2009,19(8):1707-1709.
[16]周碧青,张金彪.高效液相色谱法快速测定蜂王浆中四环素类抗生素残留[J].福建分析测试,2015,24(4):10-13.
[17]唐小兰,魏仲珊,王亮亮,等.鲜冻水产品中违禁兽药残留高效液相色谱-串联质谱法比较与改进研究[J].现代食品,2015(17):35-42.
[18]陈京都,吴红军,成强,等.液相色谱-质谱联用技术在水产品兽药残留检测中的应用[J].化学分析计量,2015,24(5):105-107.
[19]李诗言,张海琪,郑重莺,等.高效液相色谱-四极杆-飞行时间质谱法筛查中华鳖中42种兽药残留[J].中国渔业质量与标准,2015,5(1):43-51.
[20]李晓雯,迟秋池,夏苏捷,等.高效液相色谱-四极杆-飞行时间质谱法检测猪肉中22种磺胺类兽药残留[J].食品安全质量检测学报,2015,6(5):1735-1742.
[21]李丽莉,罗轶,何颂华,等.高效液相色谱-串联质谱法测定鱼肉中8种喹诺酮类药物的残留量[J].中国食品卫生杂志,2012,24(1):37-38.
[22]文一,姚瑞华,孙宏亮,等.超高效液相色谱-质谱连用法同时测定地下水中23种药物残留[J].环境科学学报,2015,35(7):2127-2135.
[23]付体鹏,张峰,刘力,等.高效液相色谱-电喷雾-iFunnel-串联质谱法同时测定猪尿液中20种禁用兽药残留[J].分析测试学报,2013,32(10):1153-1159.
[24]沈虎琴,檀华蓉,祁克宗,等.畜禽粪便中的氟喹诺酮类与四环素类兽药残留同时提取的方法研究[J].分析测试学报,2012,31(3):302-306.
[25]卜明楠,石志红,康健,等.QuEChERS结合-LC-MS/MS同时测定虾肉中72种兽药残留[J].分析测试学报,2012,31(5):552-558.
[26]曲斌.QuEChERS在动物源性食品兽药残留检测中的研究进展[J].食品科学,2013,34(5):327-331.
[27]王立琦,贺利民,曾振灵,等.液相色谱-串联质谱检测兽药残留中的基质效应研究进展[J].质谱学报,2011,32(6):321-332.
Determination of 35 Kinds of Veterinary Drugs Residues in Pork by QuEChERS Purification-UPLC-MS
LIANG Fei-yan1,ZHANG Ke-ming2,LU Ri-gang2
(1.Guangxi-Asean Center for Food and Drug Safety Control,Nanning,Guangxi 530021;2.Guangxi Institute for Food &Drug Control,Nanning,Guangxo 530021)
[Objective]To establish the QuEChERS purification-UPLC-MS detection method of 35 kinds of veterinary drugs residues in pork.[Method]The pork samples were extracted by Na2EDTA-Mcllvaine (pH 4.0) and 2.5% acetic acid acetonitrile respectively,the solution obtained was purified and concentrated by using the dehydrating agent of NaCl and anhydrous Na2SO4and the adsorbent of -NH2,and then was detected in electrospray ionization in positive ion mode with the method of multiple reaction monitoring (MRM),qualitative and quantitative analysis was conducted by matrix spiked blank curve external standard method.[Result]The linear relations of 35 kinds of veterinary drugs were good in the range of 0.5-50.0 ng/mL concentration and the correlation coefficients were greater than 0.995,the average recovery rates of 4 different concentrations were 60%-110%,relative deviations (RSD) were less than 10%,the detection limits(LOD,S/N=3∶1) were 0.02-1.20 μg/kg,the quantitative limits (LOQ,S/N=10∶1) were 0.07-4.00 μg/kg.[Conclusion]The method is simple,rapid,reliable,sensitive and efficient,and can be used for the detection and monitoring of multi drug residues in pork.
Veterinary drug residue;Detection;QuEChERS;Ultra performance liquid chromatography tandem mass spectrometry
梁飞燕(1983- ),女,广西南宁人,主管药师,硕士,从事食品、保健食品、药品的检验检测与研究。
2016-07-17
S 851.34+7
A
0517-6611(2016)25-041-05
猜你喜欢
化工管理(2022年13期)2022-12-02
能源工程(2021年1期)2021-04-13
理化检验-化学分册(2020年12期)2021-01-26
中国蜂业(2018年4期)2018-05-09
当代化工研究(2016年6期)2016-03-20
分析测试学报(2015年6期)2016-01-13
核科学与工程(2015年2期)2015-09-26
食品工业科技(2014年15期)2014-03-11
无机化学学报(2014年3期)2014-02-28
河南科技(2014年7期)2014-02-27
