
苯甲酸阿格列汀合成研究进展
2016-09-21 03:01张雷刚牟敏仁
化工生产与技术 2016年2期
张 峰,张雷刚,牟敏仁
(浙江永太科技股份有限公司,浙江 临海 317016)
苯甲酸阿格列汀合成研究进展
张 峰,张雷刚,牟敏仁
(浙江永太科技股份有限公司,浙江 临海 317016)
介绍了苯甲酸阿格列汀的药学背景,从起始原料出发总结和归纳了苯甲酸阿格列汀的现有合成路线,并对每条路线进行了分析和总结。苯甲酸阿格列汀合成的主要起始原料有6-氯尿嘧啶、6-氯-3-甲基尿嘧啶、1-(2-异氰苄基)-3-甲基脲、丙二酸单乙酯和4-甲氧基-3-氧代丁腈。认为路线以6-氯-3-甲基尿嘧啶为起始原料,反应条件温和,后处理简单,更加符合工业化生产的要求。但应进一步优化反应条件,减少副反应发生,降低成本。
苯甲酸阿格列汀;合成路线;6-氯-3-甲基尿嘧啶
苯甲酸阿格列汀化学名2-[6-[(R)-3-氨基-1-哌啶基]-3,4-二氢-3-甲基-2,4-二氧代-1(2H)-嘧啶基]甲基]苯甲腈苯甲酸盐,是日本武田公司研发的一种二肽基肽酶(DPP-4)抑制剂,临床上用于II型糖尿病的治疗[1-2]。DPP-4抑制剂主要是通过特异性结合DPP-4酶并抑制其活性,维持体内胰高血糖素样肽1(GLP-1)和葡萄糖依赖性促胰岛素多肽(GIP)的水平,促进胰岛素的分泌,从而降低血糖的目的[3-4]。苯甲酸阿格列汀作为新型的DPP-4抑制剂,具有选择性高,安全性好的特点[5-7]。该药物于2010年4月获得日本厚生劳动省的上市批准,并于2013年成功在美国和中国上市[8-9]。
苯甲酸阿格列汀的合成最初是以嘧啶环、哌啶环和苄基的拼接为主要路线,后来又相继报道了以1-(2-异氰苄基)-3-甲基脲、丙二酸单乙酯以及4-甲氧基-3-氧代丁腈为原料构建嘧啶环的合成方法。根据起始原料的不同,苯甲酸阿格列汀的合成主要归纳为5条路线。
1 以6-氯尿嘧啶为起始原料
日本武田公司(Takeda)于2005年公布了以6-氯尿嘧啶(2)为起始原料合成苯甲酸阿格列汀的方法。根据该方法,在NaH和LiBr存在下,以N,N’-二甲基甲酰胺(DMF)-二甲基亚砜(DMSO)构成的混合溶剂中用邻氰基溴苄将6-氯尿嘧啶烷基化,得到中间体4。然后中间体4与碘代甲烷(MeI)在NaH存在下于DMF-四氢呋喃(THF)混合溶剂中进一步烷基化得到1,3-二取代尿嘧啶(5)。随后化合物5在含有NaHCO3的甲醇中或含有K2CO3的含水异丙醇中加热,与3-(R)-氨基哌啶双盐酸盐(6)发生亲核取代反应,产生阿格列汀(7),阿格列汀再与苯甲酸的乙醇溶液反应得到对应的苯甲酸盐(1)。该方法的总收率为约20%~25%[10]。反应式为:

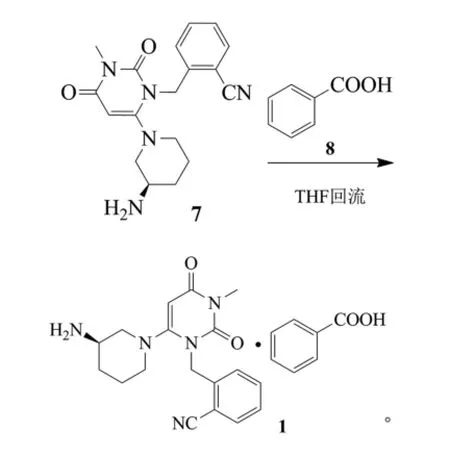
上述方法中间体2-(6-氯-3-甲基-2,4-二氧代-3,4-二氢-2H-嘧啶-1-基甲基)-苄氰(5)与(R)-3-氨基哌啶二盐酸取代的过程中,由于(R)-3-氨基哌啶二盐酸中有2个氨的活性位置,所以很容易同时与2个氨的位置发生取代而产生杂质,而且杂质不容易除去,会大大影响产品纯度,降低反应收率。此外该路线多次采用高沸点的溶剂混合物(DMSO-DMF、THFDMF),难以分离纯化进行回收利用,多次使用高危险物质氢化钠,需要无水溶剂作为反应介质,而烷基化所使用的碘甲烷为高毒性试剂,不推荐使用。
李明杰等在日本武田公司原研路线的基础上对工艺稍加改进,各部操作均在氮气保护下完成,并用3-(R)-Boc-氨基哌啶(9)代替3-(R)-氨基哌啶双盐酸盐6,提高了反应选择性,减少了步骤三中杂质的生成[11]。反应式为:
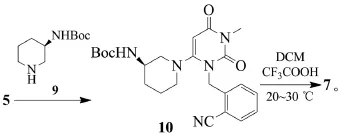
但该路线依然没有改变原研路线使用高沸点溶剂和危险物质的缺点。此外,在该改进中,化合物10在冷却的体系中大量析出甚至使体系固化,造成分离困难,严重影响产品纯度和收率,该路线总收率为14.1%。
2 以6-氯-3-甲基尿嘧啶为起始原料
Feng Jun等公开了以6-氯-3-甲基尿嘧啶(11)为起始原料的另1种苯甲酸阿格列汀的合成方法。该方法第1步采用N,N-二异丙基乙二胺为缚酸剂,化合物3和化合物11在甲苯和N-甲基吡咯烷酮组成的混合溶剂中反应得到化合物5;第2步中间体5以碳酸钾作为缚酸剂与3-(R)-氨基哌啶双盐酸盐在异丙醇和水的混合溶剂中反应得到化合物7,并最终与苯甲酸成盐得到目标产物[12]。反应式为:
该方法直接以6-氯-3-甲基尿嘧啶作原料回避了甲基化步骤,并取消了氢化钠的使用,但第2步操作仍然存在反应选择性问题。
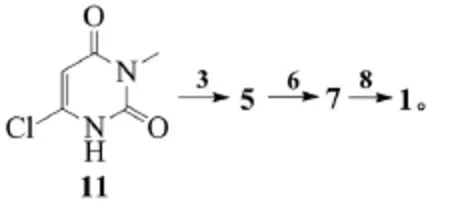
刘昭文等对上述制备苯甲酸阿格列汀的方法再次进行了改进[13]。该路线以6-氯-3-甲基尿嘧啶(11)为原料,与邻氰基溴苄(3)在三正丁胺做缚酸剂条件下反应制得2-(6-氯-3-甲基-2,4-二氧代-3,4-二氢-2H-嘧啶-1-基甲基)-苄氰(5),继续在三正丁胺条件下与(R)-3-氨基哌啶二盐酸发生取代反应,最后与苯甲酸成盐制得苯甲酸阿格列汀。该路线采用(R)-3-氨基哌啶二盐酸仍然存在反应选择性问题。
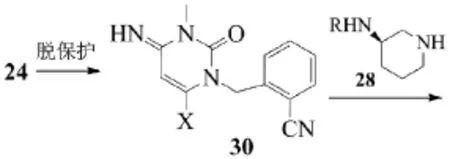
针对该条路线的不足,某公司于2013年提出了改进方案。该专利以6-氯-3-甲基尿嘧啶(11)原料,与2-氰基溴苄反应得到中间体5,再与(R)-3-Boc氨基哌啶制备得到叔丁氧羰基保护的化合物,并选用对甲苯磺酸脱去Boc保护基团,最后阿格列汀与苯甲酸成盐得到终产物[14]。反应式为:

该反应方案的总收率可达59.7%,产率更高,产品质量更好。
但该方案生成化合物10的反应中采用乙醇作溶剂,乙醇的亲核性强,可与中间体5发生亲核取代反应,产生大量副产物。高尚等人以异丙醇代替乙醇作为该反应溶剂的方法,由于异丙醇的空间位阻效应,有效避免了溶剂参与反应而生成副产物[15]。
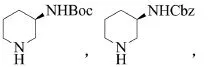
此外,针对2-氨基哌啶在构建阿格列汀母体结构时反应选择性的问题,目前已有多篇文献公开报道了对哌啶环上氨基的保护方案[16-19]。几种氨基保护后形成的中间体:

3 以1-(2-异氰苄基)-3-甲基脲为原料
原料1-(2-异氰苄基)-3-甲基脲(12)的合成:
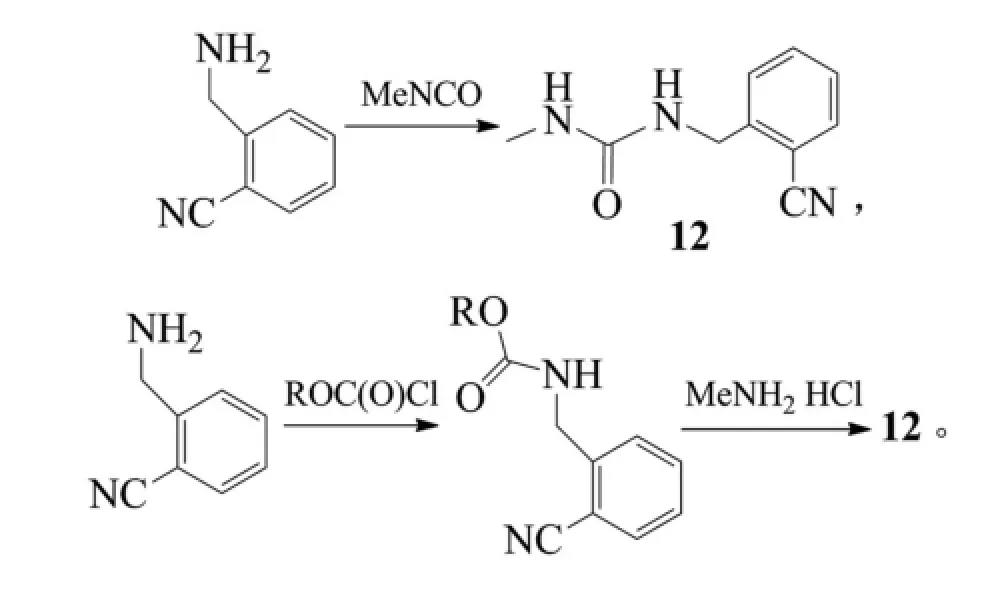
以色列MAPI医药公司公布了1种新的苯甲酸阿格列汀制备方法,该工艺需先得到1-(2-异氰苄基)-3-甲基脲(12),然后与丙二酸二乙酯反应形成三酮衍生物(13)(收率78%~85%),再用POCl3氯化得到二酮衍生物(收率75%),随后与(R)-3-Boc-氨基哌啶(9)反应得到中间体10(收率93%~96%),最后通过加入溶于乙醇的苯甲酸溶液,脱去Boc保护,析出苯甲酸阿格列汀[20]。反应式为:
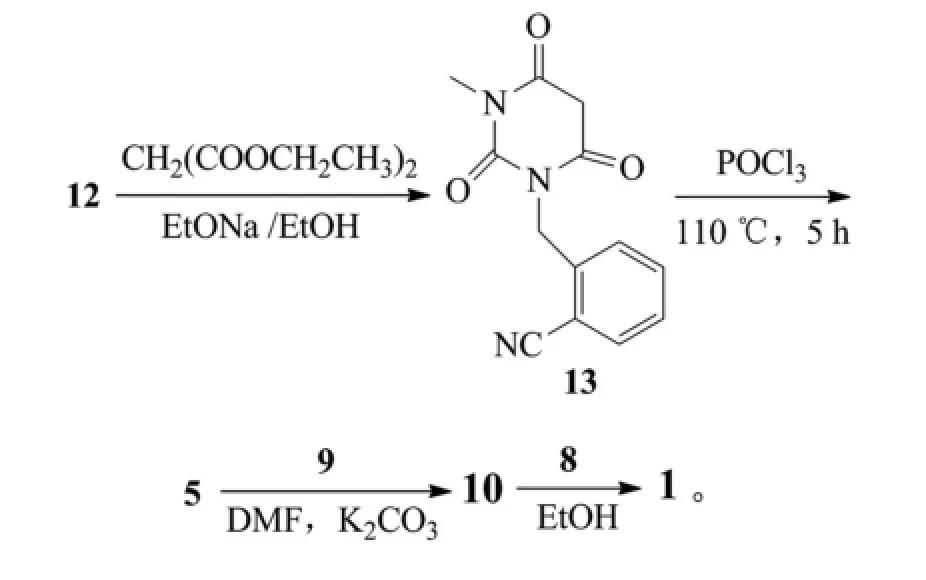
上述路线的需先制备原料12,路线较长;另外三酮衍生物13与POCl3的反应选择性差,会生成大量4-氯衍生物杂质(4-氯与6-氯衍生物摩尔比1: 1),难以除去,并影响反应收率;最后用苯甲酸脱Boc得到苯甲酸盐,由于苯甲酸的酸性较弱无法使Boc上氨基甲酸酯的氧质子化,无法失去叔丁基碳正离子,即苯甲酸几乎不可能使Boc保护基脱除,故该条路线可行性不大。
以色列MAPI医药公司的同一篇专利中还公布了另外1条合成路线:
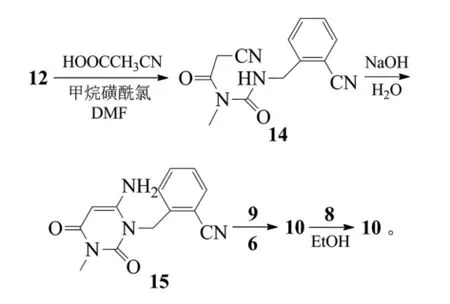
该制备方法同样存在诸多缺点。首先,原料1-(2-异氰苄基)-3-甲基脲(12)不易获得;其次,中间体14用NaOH溶液来进行水解制备中间体15,容易破坏产品,使产物纯度大大降低;最后用苯甲酸直接与叔丁氧羰基保护的化合物(10)脱Boc得到苯甲酸盐,苯甲酸酸性太弱,根本无法顺利脱下Boc,也就无法获得最终产物苯甲酸阿格列汀。
4 以丙二酸单乙酯为原料
1项专利公布了另1种苯甲酸阿格列汀的制备方法。该方法以丙二酸单乙酯(16)为原料,与氯化亚砜发生酰化反应,得到丙二酸单乙酯酰氯(17),然后将化合物17滴加到(R)-3-Boc-氨基哌啶的四氢呋喃溶液中,以N,N-二异丙基乙胺(DIEA)做缚酸剂,得到中间体18,该中间体随后在甲醇钠的甲醇溶液中与化合物12反应,得到中间体10,再通过脱Boc保护和成盐反应制得苯甲酸阿格列汀[21]。反应式为:
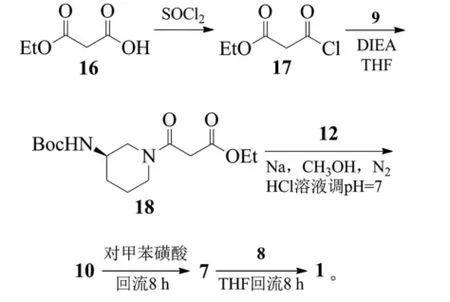
该方法在醇钠的强碱条件下自行构建嘧啶环,路线设计合理,但整条路线总收率不高。
5 以4-甲氧基-3-氧代丁腈(1919)为原料
专利WO2015092739公布了以4-甲氧基-3-氧代丁腈(19)为起始原料合成苯甲酸阿格列汀的2种新方法[22]。
方法一。4-甲氧基-3-氧代丁腈与甲基脲(20)在碱性条件下反应得到中间体21,然后对中间体21上的氨基进行保护后与邻氰基苄卤代物(23)发生亲核取代反应得到中间体24,随后再通过氨基的脱保护和氧化2步反应得到三酮衍生物(26),三酮衍生物经过一步卤化反应再与氨基保护的哌啶衍生物发生亲核取代反应生成关键中间体29,化合物29在酸性条件下脱保护形成阿格列汀,最后与苯甲酸成盐得到目标化合物。反应式为:

该条路线步骤较多,三酮衍生物的卤化存在严重的选择性问题,导致大量杂质产生;多次进行氨基的保护与脱保护,不仅操作繁琐,还大大降低反收率。
方法二。首先采用与方法一相同的手段制得中间体24,随后调整了氨基的氧化和羰基的卤化顺序,避免了三酮衍生物卤化的选择性问题。反应式为:

但整条路线仍然步骤较多,还多次涉及氨基的保护与脱保护,操作繁琐,反应总收率低,不推荐作为工艺生产的路线。
6 小结
通过对苯甲酸阿格列汀多条合成路线的综合对比分析,发现路线1.2以6-氯3-甲基尿嘧啶为原料与2-氰基溴苄反应得到中间体5,再与(R)-3-Boc氨基哌啶制备得到叔丁氧羰基保护的化合物,然后脱去保护基团并与苯甲酸成盐,整条路线工艺简便,反应选择性好,收率高,为制备苯甲酸阿格列汀的较优路线,但还可以进一步优化反应条件,减少副反应发生,降低成本。
[1]Thomas G,Volker K,Ulrich J.Griesser.Alogliptin and its benzoate salt[J].Acta Cryst,2013,C69:674-678.
[2]赵兴旺,吴冰.阿格列汀(Alogliptin)[J].中国药物化学杂志,2013,23(4):337.
[3]Lucienne J J.Dipeptidyl peptidase IV and its inhibitors: Therapeutics for type 2 diabetes and what else?[J].J Med Chem,2014,57:2197-2212.
[4]郝群,蔡正艳,周伟澄.二肽基肽酶-抑制剂构效关系研究进展[J].世界临床药物,2009,30(8):487-497.
[5]Thomas L,Eckhardt M,Langkopf E.(R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-meth-ylquinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione(BI1356),anovel xanthine-based dipeptidyl peptidase 4 inhibitor,has a superior potency and longer duration of action compared with other dipeptidyl peptidase4 inhibitor[J].J Pharmacol Exp Ther,2008,325(1):175-182.
[6]Bumsup L,Lihong S,Daniel B.Pharmacokinetic,pharmcodynamic,and efficacy profiles of alogliptin,a novel inhibitor of dipeptidyl peptidase-4,in rats,dogs and monkeys[J].Eur J Pharmacol,2008,589(1/3):306-314.
[7]Covington P,Christopher R,Daven-port M,et al.Pharmacokinetic,pharmacodynamic,and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin:a randomized,dou ble-blind,placebo-controlled,multiple-dose study in adult patients with type 2 diabetes[J].Clin Ther,2008,30(3):499-512.
[8]张淑芳.治疗糖尿病的新药NESINA获批在日本上市[J].中国执业药师,2010,12(6):541.
[9]Scott L J.Alogliptin:a review of its use in the management of type 2 diabetes mellitus[J].Drugs,2010,70(15):2051-2072.
[10]Feng J.Dipeptidyl peptidase inhibitor:EP,1586571A[P]. 2005-10-19.
[11]李明杰,刘新泉,李晓峰,等.苯甲酸阿格列汀的制备方法:中国,103193762A[P].2013-07-10.
[12]Feng J,Gwaltney S L,Stafford J A.Dipeptidyl peptidase inhibitors:WO,2007035629[P].2006-09-15.
[13]刘昭文,吴龙火,王恩军.苯甲酸阿格列汀的合成[J].海峡药学,2011,23(9):214-215.
[14]崔翼,罗邻涛,周定康,等.一种苯甲酸阿格列汀的制备工艺:中国,102942556A[P].2013-02-27.
[15]高尚,俞传明,何人宝,等.苯甲酸阿格列汀关键中间体合成工艺改进[J].浙江化工,2016,47(2):17-19.
[16]Bhatt N G,Naik S,Sharma A K,et al.Process for Preparation of Alogliptin:WO,2014188334[P].2014-11-27.
[17]赵哲,丁怀伟,赵明明,等.苯甲酸阿格列汀的合成工艺研究[J].中国医药导论,2014,16(10):1352-1353.
[18]叶天健,陈鑫,叶继华,等.一种苯甲酸阿格列汀的新制备方法:中国,103819450A[P].2014-05-28.
[19]Zhou Y X,Zhou W T,Sun L L,et al.Characterization of process-related impurities including forced degradation products of alogliptin benzoate and the development of the corresponding reversed-phase high-performance liquid chromatography method[J].J Sep Sci,2014,37:1248-1255.
[20]Marom E,Mizhiritskii M,Rubnov S.Process for the preparation of alogliptin and its salts using cyclocondensation as key step:WO,2010109468A[P].2010-09-30.
[21]莫国宁.一种苯甲酸阿格列汀的合成方法:中国,104086527[P].2014-10-08.
[22]Vadall L R,Dasari S R,Manukonda S R,et al.Process for preparation of alogliptin:WO,2015092739[P].2015-06-25.
美商务部对华氢氟烃冷媒作出反倾销初裁
2016年1月22日,美国商务部对华氢氟烃制冷剂作出反倾销初裁:裁定山东东岳化工有限公司等7家出口商/生产商的倾销幅度为92.88%;浙江蓝天环保氟材料有限公司等8家出口商/生产商的倾销幅度为91.99%;大金氟化工(中国)有限公司等10余家出口商/生产商的倾销幅度为92.60%;中国普遍的倾销幅度为210.46%。
2015年7月16日,美国商务部宣布,对原产于中国的进口氢氟烃制冷剂启动反倾销调查。涉案产品海关编码为3824.78.0000、2903.39.2030。(本刊编辑部)
巨化10 kt/a HFC-245fa项目一期
巨化集团子公司浙江衢州巨新氟化工有限公司10 kt/a HFC-245fa(一期)项目已具备开工条件,工程建设地址位于巨化集团公司内,投资估算约500万元人民币,建设周期约150天。
项目建成后将形成10kt/a HFC-245fa的生产能力,项目分2期实施,其中一期建设5kt/a HFC-245fa,二期建设5 kt/a HFC-245fa。
HFC-245fa最主要的用途是作为物理发泡剂用于硬质聚氨酯泡沫塑料的发泡,由其制得的发泡剂性能相比于CFC-11和HCFC-141b有所提高,且HFC-245fa不燃烧爆炸,相比戊烷类发泡剂更加安全,是一种理想的替代HCFC-141b的第3代发泡剂。此外,随着研究的不断深入,HFC-245fa的应用范围也越来越广,在制冷行业中可以单独或与其他组分混配用作制冷剂,还可以用于制备清洁剂、灭火剂、气雾剂、膨胀剂等。
目前我国大部分小家电、热水器和小部分冷柜企业仍在使用HCFC-141b作为发泡剂,这些企业都将面临发泡剂的替代问题,其替代品主要有环戊烷和HFC-245fa。环戊烷具有易燃易爆性,而HFC-245fa不燃,且可以降低泡沫密度,节约发泡材料,提高泡沫质量此外,与普通的环戊烷发泡剂相比,HFC-245fa发泡剂可以使冰箱节能8%,可以有效协助家电企业突破环戊烷泡沫隔热保温效果的瓶颈,使产品达到更高的节能水平。2010年,HFC-245fa在我国的销量翻了4~5倍,预计今后将有更好的市场前景。
项目的原料之一四氯化碳是甲烷氯化物装置的副产物,其对臭氧层有破坏作用,是一种ODS,国家环保部已于2008年停止四氯化碳作为生产氟利昂的原料及作为助剂和清洗剂的使用,大量甲烷氯化物装置的副产四氯化碳面临出路问题。该项目的实施,对于公司平衡四氯化碳产品,保护臭氧层具有重要的现实意义。(本刊编辑部)
综述
TQ463+.54
A
10.3969/j.issn.1006-6829.2016.02.008
2016-01-15;
2016-02-04
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
精神医学杂志(2021年2期)2021-06-23
民间文学(2021年11期)2021-03-31
民间文学(2020年8期)2020-08-31
国外医药(抗生素分册)(2016年1期)2016-07-10
广东第二课堂·小学(2016年6期)2016-05-14
中国继续医学教育(2015年5期)2016-01-07
中国继续医学教育(2015年3期)2016-01-06
中外医疗(2015年11期)2016-01-04
中国药理学通报(2014年2期)2014-05-09
