
利福昔明的合成研究
2016-09-13 07:19:15余丽华张海梁王宇光浙江工业大学生物工程学院浙江杭州310014
浙江化工 2016年8期
余丽华,张海梁,袁 龙,王宇光(浙江工业大学生物工程学院,浙江 杭州 310014)
利福昔明的合成研究
余丽华,张海梁,袁龙,王宇光
(浙江工业大学生物工程学院,浙江杭州310014)
以利福霉素S、液溴、2-氨基-4-甲基吡啶、抗坏血酸(VC)为主要原料,经过溴化、环合、还原三个步骤制备了利福昔明。结果表明,最优工艺条件为:溴化反应采用乙醇和水溶剂体系,利福霉素S与溴的摩尔比为1∶1.2;环合反应中3-Br-利福霉素S与2-氨基-4-甲基吡啶的摩尔比为1∶1.6,反应温度为63℃。在该优化条件下,产品总收率达85.1%。
利福昔明;合成;工艺优化
利福昔明(rifaximin)是利福霉素SV的半合成衍生物,属于广谱肠道抗生素。它抑制细菌蛋白质的合成,从而呈现杀菌作用,对多种革兰阳性菌、革兰阴性及厌氧菌均有高度抗菌活性。且由于本药在肠道几乎不被吸收,副作用小,利福昔明经常用作为一种局部肠道治疗药物[1]。该药在肠道感染、肝性脑病、憩室病、溃疡性结肠炎、肠道内细菌过度生长、肠内胀气等相关疾病中的应用日趋增加,疗效较好[2-10]。
由于利福昔明具有如此优越的临床应用,因而通过有效的方法合成利福昔明受到了学术界的普遍关注。目前来说,利福昔明的合成路线主要有[11-13]:①以利福霉素B为原料,碘作为催化剂和2-氨基-4-甲基吡啶的反应,接着用维生素C还原,得到利福昔明,产率为67%;②以利福霉素S或利福霉素O为原料,碘作为催化剂与2-氨基-4-甲基吡啶的反应,再与维生素C反应制备利福昔明,产率约为78%。但已报道的方法具有收率低、工业化生产成本较高、提取工艺困难等缺点。本文的合成路线如图1所示。以利福昔明S 1和溴为原料合成3-溴-利福霉素S 2,再与2-氨基-4-甲基-吡啶环合,反应生成环合产物3,接着通过抗坏血酸(VC)将上一步的产物酸化还原得到利福昔明4。
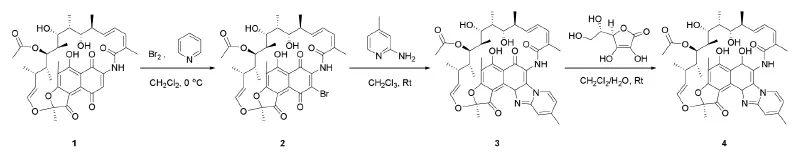
图1 利福昔明合成路线
1 实验部分
1.1试剂与仪器
利福霉素S,工业品;所用试剂均为分析纯;1HNMR由瑞士 Bruker公司 Bruker Avance-400 MHz核磁共振仪测定;有机反应用薄层硅胶板跟踪,紫外灯检测。
1.2中间体2(3-Br-利福霉素S)的合成(溴化反应)
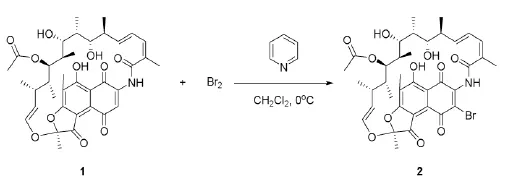
先将吡啶(960 μL,12 mmol)、二氯甲烷(20 mL)加入50 mL单口圆底烧瓶中,向其中逐滴加入Br2(12 mmol,624 μL),待用。在500 mL的圆底烧瓶中加入利福霉素S(6.9575 g,10 mmol)和二氯甲烷(200 mL),搅拌,使其完全溶解。在冰浴条件下,向其中滴加上述吡啶和液溴的反应液,搅拌,用TLC跟踪反应进程,展开剂为EtOAc-PE。反应4 h到达终点后,将反应液加入1.5 L的大烧杯中,向其中依次加入100 mL冰乙酸,1000 mL水,静置,有大量淡黄色固体析出,抽滤,固体用纯净水洗涤2~3次,干燥得3-Br-利福霉素S 7.0269 g,产率:92.8%,可直接用于下一步反应。
1H NMR(400 MHz,CDCl3)δ 12.93(s, 1H),7.20(s,1H),6.37(d,J=12.8 Hz,1H),5.24(d,J=10.5 Hz,1H),4.98(dd,J=12.8,10.1 Hz,1H),4.56(dd,J=10.5,2.1 Hz,1H),4.27~4.18(m,1H),3.54(d,J=10.5 Hz,1H),3.26(dd,J=10.1,3.1 Hz,1H),3.17(d,J= 4.4 Hz,1H),3.13(s,3H),2.90(s,1H),2.80 (d,J=8.7 Hz,1H),2.35(s,3H),2.32~2.23 (m,1H),2.19(ddd,J=10.5,7.2,3.1 Hz,1H),2.09(s,3H),1.99(s,3H),1.68(s,3H),1.05(d,J=6.9 Hz,3H),0.93(d,J=7.0 Hz,3H),0.67(d,J=6.9 Hz,3H),0.34(d,J=7.2 Hz,3H)。
1.3中间体3的合成(环合反应)
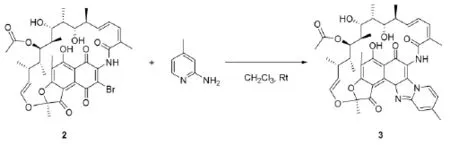
取3-Br-利福霉素S(5 mmol,3.8737 g)加入250 mL的圆底烧瓶中,用适量三氯甲烷完全溶解,再加入稍过量的2-氨基-4-甲基吡啶(8 mmol,0.864 g),在常温下搅拌,用TLC跟踪反应进程,展开剂为EtOAc-PE。反应5 h到达终点后,有机层用2 mol/L的盐酸洗涤除去过量的2-氨基-4-甲基吡啶,再用蒸馏水洗涤有机相,使pH保持在6~7左右,用无水硫酸镁干燥,浓缩,得到未酸化的利福昔明3.5617 g,产率:92.6%。
1H NMR(400 MHz,CDCl3)δ 13.24(s,1H),9.29(d,J=6.9 Hz,1H),7.79(s,1H),7.00(d,J=7.0 Hz,1H),6.62(dd,J=13.6,5.2 Hz,2H),6.52(t,1H),6.29(t,1H),5.36 (dd,J=12.4,6.2 Hz,1H),5.18(t,1H),4.79 (d,J=10.4 Hz,1H),4.01(s,1H),3.57(d,J =6.6 Hz,1H),3.46(s,1H),3.08(s,3H),2.92(d,J=9.8 Hz,1H),2.86(d,J=6.8 Hz,1H),2.52(s,3H),2.27(s,3H),2.23(s,3H),2.10(s,1H),2.04(s,3H),1.91(t,1H),1.84(t,1H),1.76(s,3H),1.72(t,1H),1.38 (t,3H),1.25(t,3H),1.08(t,J=6.7 Hz, 1H),0.92(t,3H),0.53(t,J=6.8 Hz,3H)。
1.4产物4(利福昔明)的合成(还原反应)
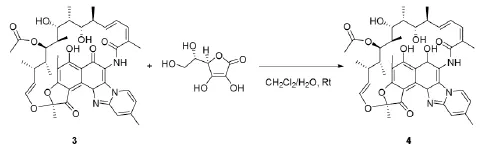
取未酸化的利福昔明 (2 mmol,1.5675 g),用适量二氯甲烷溶解,在常温搅拌状态下滴加抗坏血酸(3 mmol,0.528 g,VC),其中抗坏血酸用少量水溶解,用TLC跟踪反映进程,展开剂为CH2Cl2-MeOH。反应1 h到达终点后,水洗两到三次,用无水硫酸镁干燥,浓缩有机相,干燥得目标产物利福昔明成品1.4965 g,产率:97.0%。
1H NMR(400 MHz,CDCl3)δ 13.25(s,1H),9.31(d,J=6.9 Hz,1H),7.80(s,1H),7.00(d,J=7.0 Hz,1H),6.51(dd,J=13.6,5.2 Hz,1H),6.36(t,1H),6.25(t,1H),5.36 (dd,J=12.4,6.2 Hz,1H),4.81(d,J=10.4 Hz,1H),4.02(s,1H),3.58(d,J=6.6 Hz,1H),3.46(s,1H),3.12(s,1H),3.09(s,3H),2.95(d,J=9.8 Hz,1H),2.88(d,J=6.8 Hz,1H),2.53(s,3H),2.28(s,4H),2.25(s,3H),2.05(d,J=5.7 Hz,6H),1.28(dd,J= 16.5,9.0 Hz,3H),1.02(d,J=6.9 Hz,2H),0.93(d,J=7.0 Hz,3H),0.85(d,J=6.8 Hz,2H),0.67(d,J=6.7 Hz,1H),0.55(d,J=6.7 Hz,3H),0.21(d,J=7.0 Hz,1H)。
2 结果与讨论
2.1溴化反应的条件优化
2.1.1原料摩尔比对溴化反应产率的影响
首先考察了利福霉素S与液溴的摩尔比对反应收率的影响,结果如图1所示。结果显示摩尔比小于1∶1.2时,产物收率能够达到最大,因此我们选用最节省原料的1∶1.2。
2.1.2溶剂对溴化反应的影响
接着考察了多组溶剂对产率的影响。结果发现:在相同反应条件下,乙醇/水的反应效果能达到最佳。在此基础上,我们继续对不同比例的乙醇/水溶剂体系对反应产率的影响进行研究,结果如图2所示。
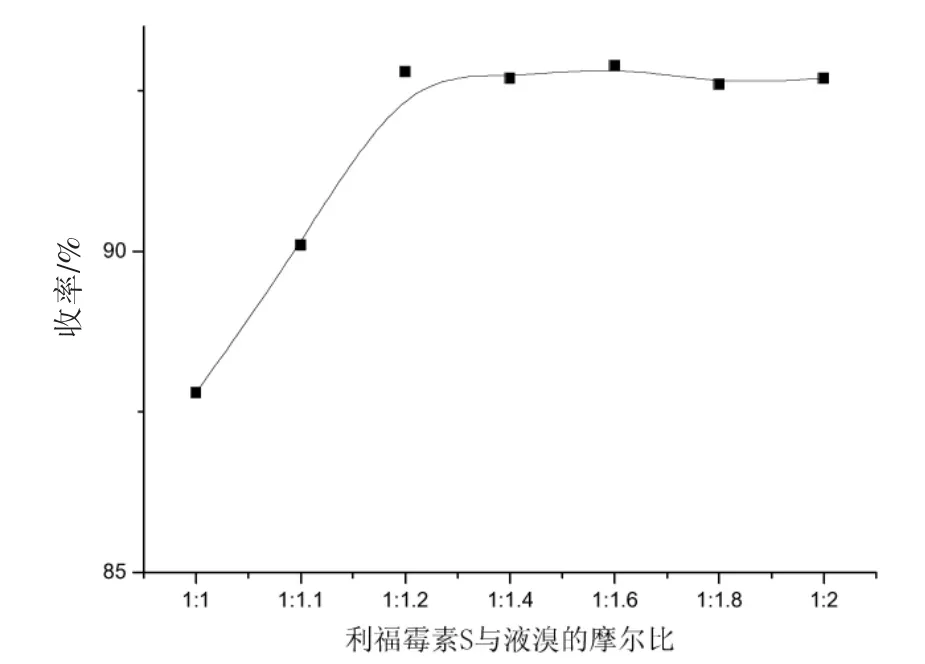
图1 原料摩尔比对反应产率的影响
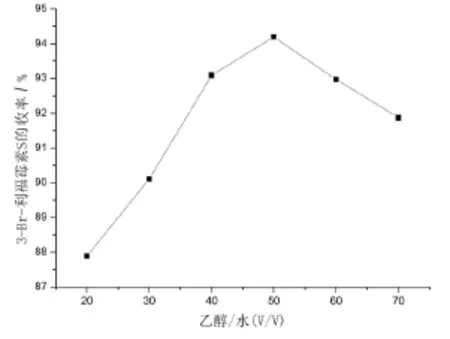
图2 不同比例的乙醇/水对反应产率的影响
与二氯甲烷相比,V/V=40/60、50/50、60/40的乙醇/水溶剂体系皆使得反应产率提高,其中V/ V=50时产率提高最多,达到94.2%(反应条件同1.2节)。在此反应中,选择V/V=50/50的乙醇/水溶剂体系的主要原因为:(1)原料利福霉素S溶解于水,易溶于无水乙醇;(2)水虽然会增强溶剂对原料的溶解性,但是它的存在会导致溴的消耗。
2.2对环合反应的条件优化
2.2.1原料摩尔比对环合反应的影响
首先考察了3-Br-利福霉素S与2-氨基-4-甲基吡啶的摩尔比对反应收率的影响,结果如图3所示。结果显示摩尔比小于1∶1.6时,产物收率能够达到最大,因此我们选用最节省原料的1∶1.6。
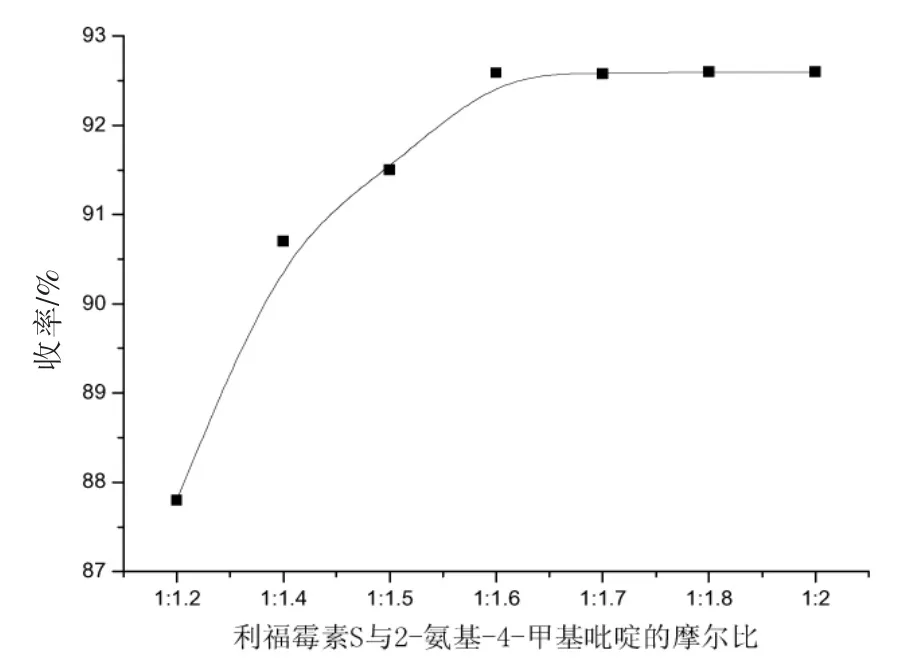
图3 原料摩尔比对反应产率的影响
2.2.2温度对环合反应的影响
接着考察了不同温度下反应对产率的影响,结果如图4所示。
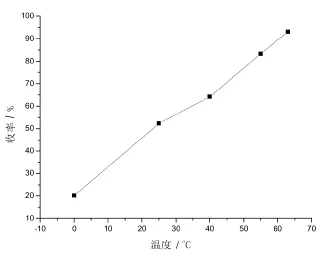
图4 温度对反应产率的影响
发现在三氯甲烷回流(63℃)条件下,相比0℃、25℃、40℃、55℃,反应产率最佳,能达到93.1%。分析主要原因是此环合反应属于吸热反应,提高温度有利于反应正向进行。
3 结论
以利福霉素S为起始原料,经过液溴溴化,接着与2-氨基-4-甲基吡啶环合,最后用抗坏血酸还原三个步骤合成得到利福昔明。本工艺路线原料易得、操作简便、工艺稳定、收率较高,易于工业化,其较佳工艺条件为:利福霉素S与溴发生反应的最佳溶剂为乙醇/水体系,比例为50/50(V/ V),且原料利福霉素S与溴的摩尔比为1∶1.2;3-Br-利福霉素S与2-氨基-4-甲基吡啶环合反应的最佳反应温度为63℃,且原料的摩尔比为1∶1.2。在该优化条件下,最终产品产率可达到85.1%,纯度为98.7%。产品结构经1H NMR确认为利福昔明。
[1] Feret B,Barner B.Rifaximin:a nonabsorbable,broadspectrum antibiotic for reduction in the risk for recurrence of overt hepatic encephalopathy[J].Formulary,2010,45 (7):210-216.
[2] Baker D E.Rifaximin:a nonabsorbed oral antibiotic[J]. Rev.Gastroenterol.Disord.,2005,5(1):19-30.
[3]Layer P,resen V.Review article:rifaximin:a minimally absorbed oral antibacterial,for the treatment of travellers’diarrhoea[J].Aliment.Pharmacol.Ther.,2010,31(11):1155-1164.
[4]Pimentel M,Park S,Mirocha J,et al.The effect of an nonabsorbed oral antibiotic(rifaximin)on the symptoms of the irritable bowel syndrome:a randomized trial[J].Ann. Inern.Med.,2006,145:557-563.
[5]Lembo A,Zakko S F,Ferreira N L,et al.Rifaximin for the treatment of diarrhea-associated irritable bowel syndrome:short term treatment leading to long term sustained response[J].Gastroenterology,2008,134(4):A545.
[6]Kimberly L,Laura H,Johnson H,et al.A combination of rifaximin and neomycin is most effective in treating irritable bowel syndrome patients with methane on lactulose breath test[J].Clin.Gastroenterol.,2010,44(8):547-550.
[7]徐大海,张楠,高金莹,等.肝性脑病治疗进展[J].临床肝胆病杂志,2011,27(2):218-221.
[8]Fritz E,Hammer H F,Lipp R W,et al.Effects of lactulose and polyethylene glycol on colonic transit[J].Aliment. Pharmaeol.Ther.,2005,21(3):259-268.
[9]Williams R,James O F,Wames T W,et al.Evaluation of the efficacy and safety of rifaximin in the treatment of hepatic encephalopathy:a double-blind randomized,dosefinding multicentre study[J].Eur.J.Gastroenterol.&Hepatol.,2000,12(2):203-208.
[10]Miglio F,Valpiani D,Rossellini S R,et al.Rifaximin,a non-absorbable rifamycin,for the treatment of hepatic encephalopathy.A double-blind,randomized trial[J].Curr. Med.Res.Opin.,1997,13(10):593-601.
[11]ALFA F S.Process for the synthesis of pyrido-imidazo rifamycins:US,4557866A[P].1985-04-26.
[12]ALFA F S.Process for the synthesis of pyrido-imidazo rifamycins:CA,1215976A1[P].1986-12-30.
[13]ALFA F S.Process for the synthesis of pyrido-imidazo rifamycins:CA,1218650A1[P].1985-09-25.
Study on the Synthesis of Rifaximin
YU Li-hua,ZHANG Hai-liang,YUAN Long,WANG Yu-guang
(College of Biologycal Engineering,Zhejiang University of Technology,Hangzhou,Zhejiang 310014,China)
In this paper,rifaximin was prepared with rifamycin S,bromine,2-amino-4-methylpyridine and vitamin C as the main raw material,via three steps of bromination,cyclization and reduction.The result showed that the optimum reaction conditions were determined as follows:In reduction,the solvent was ethanol/water and the molar ratio of rifamycin S to bromine of 1∶1.2.In cyclization,the molar ratio of rifamycin S to 2-amino-4-methyl-pyridine of 1∶1.6 and the reaction temperature is 60℃.Under the optimum condition,the yield of the product reached 85.1%.
rifaximin;synthesis;process optimization
1006-4184(2016)8-0021-05
2016-02-24
余丽华(1993-),女,硕士研究生。
王宇光(1972-),男,内蒙古包头人,副教授,博士,研究方向为药物合成。E-mail:Yuguangw@zjut.edu.cn。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
现代畜牧科技(2021年9期)2021-10-13 06:38:40
河北画报(2020年10期)2020-11-26 07:20:56
化工学报(2020年4期)2020-05-28 09:25:24
今日农业(2019年11期)2019-08-13 00:49:02
英美文学研究论丛(2018年2期)2018-08-27 01:56:44
精细石油化工(2015年3期)2015-12-14 09:07:42
无机化学学报(2014年7期)2014-02-28 17:32:28
江西理工大学学报(2013年1期)2013-03-20 14:57:07
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:40
