
基于OpenCL的蛋白质转换算法
2016-09-10 08:23:41徐沛彦施慧彬
计算机与数字工程 2016年8期
徐沛彦 施慧彬,2
(1.南京航空航天大学计算机科学与技术学院 南京 211106)(2.软件新技术与产业化协同创新中心 南京 211106)
基于OpenCL的蛋白质转换算法
徐沛彦1施慧彬1,2
(1.南京航空航天大学计算机科学与技术学院南京211106)(2.软件新技术与产业化协同创新中心南京211106)
为了提高蛋白质转换算法的加速比,提出了基于OpenCL的蛋白质转换算法。通过研究笛卡尔积算法中的并行性,论文首先在CPU中实现了串行蛋白质转换算法,再使用OpenCL技术在GPU中实现并行蛋白质转换算法。测试结果表明,使用GPU平均加速比为1.42x。
蛋白质; 转换算法; 笛卡尔积; OpenCL
Class NumberTP301.6
1 引言
蛋白质在生物体和细胞的生命过程中占有相当重要的作用[1~2]。在治疗疾病中,蛋白质为药物作用的靶标,将基因密码知识和蛋白质的结构的相关信息结合在一起,药物设计者可以设计出小分子化合物,抑制与疾病相关的蛋白质,进而达到治疗疾病的目的[3~5]。而当我们知道并需要某些与抑制疾病相关的特定的蛋白质时,蛋白质水平的高效表达是相当重要的。如何有效地得到这些特殊蛋白质将是我们需要考虑的问题。由分子生物学中心法则可知蛋白质是从RNA翻译而来。蛋白质的结构决定了蛋白质表达的功能[6]。虽然在生物学上蛋白质序列是不可能发生转换成mRNA序列的过程。由于mRNA负责传输相关遗传信息,且mRNA序列的排列顺序决定了氨基酸的排列顺序[7]。mRNA对蛋白质的结构起着重要的作用。因此从生物信息学的角度来说本文研究从蛋白质序列转换成mRNA序列可作为药物设计或生物研究的参考的基础工作且具有一定的意义。
目前可以得到的来自大量组织的完整的基因序列使得对所有基因产物的结构化和功能化的分析有需求的目标[8]。然而由于宿主的翻译系统和蛋白质编码基因序列之间的差异使得在替代的宿主上产生充足的目标蛋白质是具有挑战性的[9]。目前已经有一些方法试图获取高产量,包括使用强转录启动子[10],从宿主中删除蛋白酶编码基因[11],降低温度促使蛋白质折叠[12]。然而并没有建立一个黄金标准,和对于优化基因表达策略在不同基因之间是有差异的。往往是通过反复实验决定的。在众多被列出引出阻止有效地异源蛋白质复制因素主要有偏好密码子使用,基因产物毒性,溶解性,二级结构,mRNA稳定性[13]。Jun Yin等针对原核生物大肠杆菌提出了一种可以提升大肠杆菌中的异源蛋白质表达水平的转换方法[14]。该文献中简化了遗传密码表从而降低了转换的复杂度。该文献是用串行方法实现且用于转换的蛋白质序列长度为10。目前没有人基于笛卡尔积的思想并行的实现蛋白质转换方法。本文描述了如何在异构平台下实现并行蛋白质转换算法。实验结果和传统的CPU实现的串行蛋白质转换算法的运行时间做了对比。并行蛋白质转换算法的实现有利于算法的加速。
2 OpenCL编程
2.1OpenCL简介
OpenCL(Open Computing Language)是一种工业标准框架。它用于在配备异构计算设备的系统中开发可移植的并行应用程序。OpenCL广泛地支持各种平台,如CPU(多核处理器)、AMD GPU(图形处理器)、NVIDIA GPU(图形处理器)、移动平台。有关OpenCL的详细介绍可参考文献[15~18]。
2.2OpenCL编程步骤
1) 获取可用平台信息;
2) 获取平台上可用的设备列表;
3) 为GPU设备创建上下文环境;
4) 为GPU创建命令队列,用于宿主机将命令发送到设备;
5) 确定设备所需处理数据的大小,根据这些参数创建所需的内存对象;
6) 创建OpenCL程序对象;
7) 创建OpenCL内核对象;
8) 设置内核参数;
9) 执行内核函数;
10) 返回结果。
3 并行蛋白质转换算法
3.1算法设计
蛋白质转换算法是将蛋白质序列中的每个氨基酸按照遗传密码表中相应的三个核苷酸的组合替换,从而得到所有可能的mRNA序列。本文将基于笛卡尔积的思想进行并行蛋白质序列转换。首先将蛋白质序列用于替换相应氨基酸的密码子看作一个集合,例如A={GCU,GCC,GCA,GCG},C={UGU,UGC},在蛋白质数据库GenBank[19~20]中截取了HTA1的蛋白质序列片段为例,长度为10。蛋白质序列片段为RITPRHLQLA。查询遗传密码表并按照笛卡尔积算法,最终得到995328种不同的mRNA序列。比如mRNA序列CGUAUUACUCCUCGUCAUUUACAAUUAGCU。从得到的所有mRNA序列可以分析出蛋白质序列并行转换方法主要分为如下过程:
1) 集合中每个元素在每一次循环中重复出现的总次数p和位于该集合之后的每个集合总的元素个数有关。比如集合H中每个元素每一次重复的次数和集合L,集合Q,集合L,集合A有关。p的值为其后面四个氨基酸对应的集合的总元素个数的乘积,即p的值为288。以此类推。
2) 在第1)个过程之后每个集合需要循环的总次数q为自身集合的总元素个数和该集合之前的所有集合的总元素个数有关。比如集合I的循环次数和自身集合,集合R有关。q的值为集合I和集合R的总元素个数的乘积,即q的值为18。以此类推。
从上述分析过程中可看出并行转换方法的整个过程只需要两层循环。在两层循环之前只需要计算每次循环中集合中元素重复出现的次数p和每个集合需要循环的总次数q。而p和q的计算是完全并行的。最终可完成矩阵的填充。
3.2算法步骤
1) 并行计算集合i中每次循环中每个元素重复出现的次数p;
2) 并行计算集合i需要循环的总次数q;
3) 并行进行两层循环操作,若最外层集合i需要循环的总次数不等于q且最里层集合i中每个元素重复出现次数不为p时,将集合i中元素填充到矩阵的相应列中。
以长度为10的蛋白质序列为例,可使用一维NDRange并行处理,若蛋白质序列长度超过了用一维NDRange所能并行处理的大小,则可使用二维NDRange。工作组的大小为10,每个工作组包含一个工作项。每个工作项负责矩阵中一列的填充,矩阵中的每列的元素均来源于相应的集合中的元素。
4 实验结果
本文使用的测试平台的CPU型号为A10-6800K,主频为4.1GHz,内存大小为32G,显卡型号为HD 8670D。测试系统为Win7。用于测试的蛋白质序列长度在5~40之间,本文对于不同长度的蛋白质序列运用并行蛋白质转换算法在CPU中的运行时间和GPU中的运行时间进行了测试比对,测试结果如图1所示。
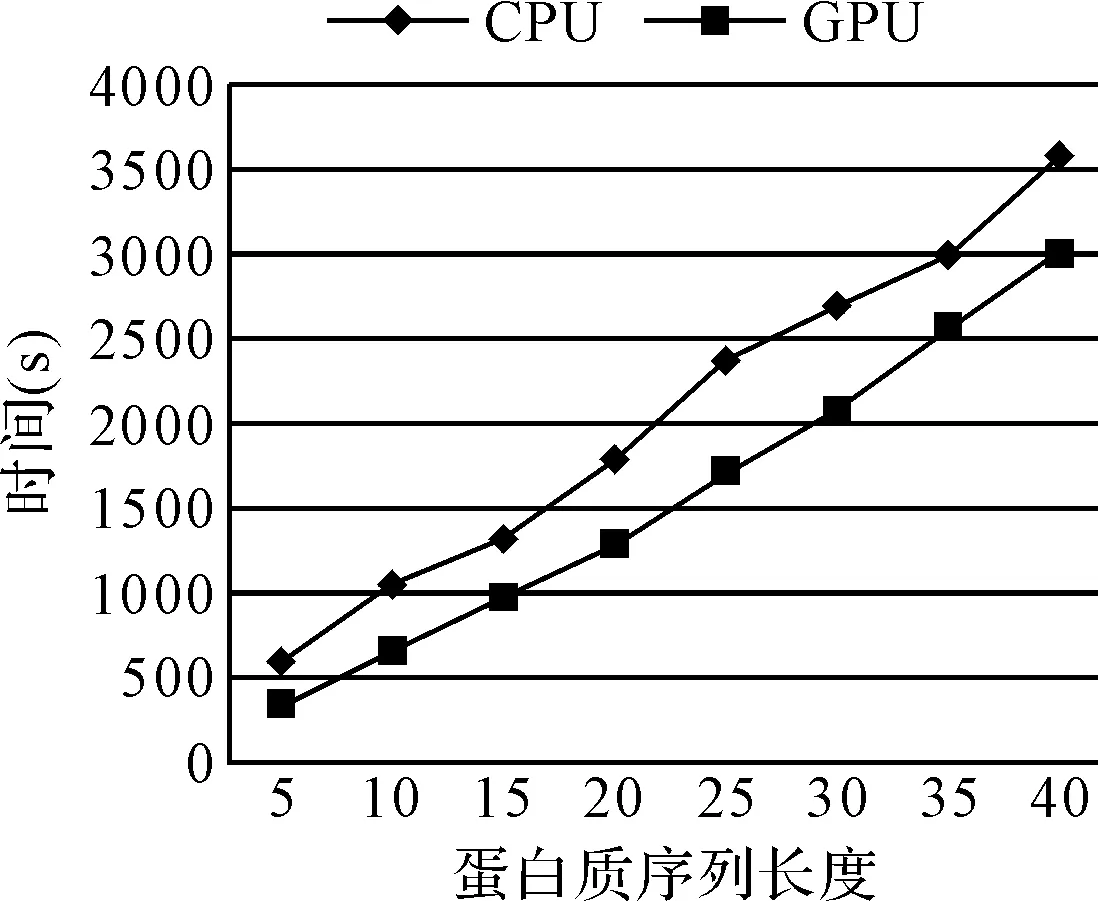
图1 测试结果
在上述测试中,图1是在不同长度的蛋白质序列下并行蛋白质转换算法在GPU上运行的时间和串行蛋白质转换算法在CPU上运行的时间。从图1中可以看出随着蛋白质序列长度的增加,无论是在GPU上运行时间还是在CPU上运行时间都会随之增加。对于并行蛋白质转换算法来说,蛋白质序列长度的增加会使得所有可能的mRNA序列的总数目增加。因此也增加了并行计算的时间。虽然在GPU上是并行执行转换操作,但是最终传回到主机内存中的数据量也随之在增加,这样就增加了数据的传输开销。因此在运行时间上会有一定的提升。对于串行蛋白质转换算法的运行时间的增加随着蛋白质序列长度的增加而增加是很显然的。但是相比于串行蛋白质转换算法上的运行时间来说,已经得到了加速。并行蛋白质转换算法在运行时间上明显优于串行蛋白质转换方法。并行蛋白质转换算法平均加速了1.42x。
5 结语
本文描述了使用OpenCL实现蛋白质并行转换算法。对串行蛋白质转换方法和并行蛋白质转换方法对不同的蛋白质序列长度下的运行时间进行了比较。虽然在异构平台下会存在数据传输开销等因素使得运行时间成本增加,但是通过测试结果依然可以看出使用OpenCL技术实现的转换方法进一步提高了算法加速比。
[1] 于大钊.基于离散小波变换的蛋白质相似性分析[D].长春:吉林大学,2013.
YU Dazhao. Proteins Similarities Analysis Base on Discrete Wavelet Transform[D]. Changchun: Jilin University,2013.
[2] 梁刚锋.蛋白质二级结构的建模与预测[D].长沙:国防科技技术大学,2005.
LIANG Gangfeng. Research on the Models of Protein Secondary Structure Prediction[D]. Changsha: National University of Defense Technology,2005.
[3] 陈宇.一个蛋白质去折叠可视化系统的设计与实现[D].苏州:苏州大学,2009.
CHEN Yu. The Design and Implementation of A Protein Unfolding Visualization System[D]. Suzhou: Soochow University,2009.
[4] 刘琨.近场表面增强拉曼散射实验技术与血清分析的研究[D].大连:大连理工大学,2008.
LIU Kun. Near-field SERS technique and its analysis in serum[D]. Dalian: Dalian University of Technology,2008.
[5] 苏洁.蛋白质分子结构的非真实感可视化研究[D].杭州:浙江大学,2006.
SU Jie. Non-photorealistic Visualization of Protein Molecules[D]. Hangzhou: Zhejiang University,2006.
[6] 陈俊.蛋白质结构预测中若干问题的研究[D].上海:上海交通大学,2013.
CHEN Jun. Studies on several problems of protein structure prediction[D]. Shanghai: Shanghai Jiao Tong University,2013.
[7] 向其林.基于结构特征与伪氨基酸组成的蛋白质结构类预测研究[D].长沙:湖南大学,2012.
XIANG Qilin. Incorporating secondary features into the general form of Chou’s PseAAC for predicting protein structural class[D]. Changsha: Hunan University,2012.
[8] Xiaoqiu Wu, Hans Jornvall, Kurt D. Berndt, et al. Codon optimization reveals critical factors for high level expression of two rare codon genes in Escherichia coli: RNA stability and secondary structure but not tRNA abundance[J]. Biochemical & Biophysical Research Communications,2004,313(1):89-96.
[9] Miyuki T, Kentaro M. Directed evolution study unveiling key sequence factors that affect translation efficiency in Escherichia coli[J]. Journal of Bioscience and Bioengineering,2013,116(5):540-545.
[10] William F S. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system[J]. Journal of Molecular Biology,1991,219(1):37-44.
[11] Grodberg J, Dunn J J. ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification[J]. Journal of Bacteriology,1988,170(3):1245-1253.
[12] Schein C H. Production of Soluble Recombinant Proteins in Bacteria[J]. Nature Biotechnology,1989,7(11):1141-1149.
[13] Jana S, Deb J K. Strategies for efficient production of heterologous proteins in Escherichia coli[J]. Applied Microbiology & Biotechnology,2005,67(3):289-298.
[14] Jun yin, Hong Tian, Lichen Bao, et al. An alternative method of enhancing the expression level of heterologous protein in Escherichia coli[J]. Biochemical and Biophysical Research Communications,2014,455:198-204.
[15] 詹云,赵新灿,谭同德.基于OpenCL的异构系统并行编程[J].计算机工程与设计,2012,33(11):4191-4195.
ZHAN Yun, ZHAO Xincan, TAN Tongde. Parallel programming of heterogeneous system based on OpenCL[J]. Computer Engineering And Design,2012,33(11):4191-4195.
[16] Benedict R G, Howes L, Kaeli D, et al. OpenCL异构计算[M].张云泉,张先轶,龙国平,等译.北京:清华大学出版社,2012:15-149.
Benedict R G, Howes L, Kaeli D, et al. Heterogeneous Computing with OpenCL[M]. Zhang Yunquan, Zhang Xianyi, Long Guoping, et al. Beijing: Tsinghua University Press,2012:15-149.
[17] Scarpion M. OpenCL in Action[M]. USA: Manning Publications,2011:3-234.
[18] Munshi A, Gaster B, Mattson T G, et al. OpenCL Programming Guide[M]. Boston: Addison-Wesley Professional,2011:8-12.
[19] Baxeyanis D A, Francis B F.生物信息学-基因和蛋白质分析的实用指南[M].李衍达,孙之荣,译.北京:清华大学出版社,2000:16-32.
Baxeyanis D A, Francis B F. Bioinformatics: a practical guide to the analysis of genes and proteins[M]. Li Yanda, Sun Zhirong. Beijing: Tsinghua University Press,2000:16-32.
[20] 王哲.生物信息学概论[M].西安:第四军医大学出版社,2002:2-104.
WANG Zhe. Fundamental Concepts of Bioinformatics[M]. Xi’an: The Fourth Military Medical University Press,2002:2-104.
Alternative Algorithm of Protein Based on OpenCL
XU Peiyan1SHI Huibin1,2
(1. College of Computer Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing211106)
(2. Collaborative Innovation Center of Novel Software Technology and Industrialization, Nanjing211106)
In order to accelerate the alternative algorithm of protein, this thesis proposes the alternative algorithm of protein based on OpenCL. Through researching the potential parallel of Cartesian product, this thesis firstly realizes the alternative algorithm of protein on CPU, then realizes parallel alternative algorithm of protein on GPU. Test results show that the average speedup of using GPU is 1.42x.
protein, alternative algorithm, Cartesian product, OpenCL
2016年2月15日,
2016年3月20日
徐沛彦,女,硕士,研究方向:计算机系统结构。施慧彬,男,博士,副教授,研究方向:计算机系统结构,可重构计算,编译优化技术。
TP301.6
10.3969/j.issn.1672-9722.2016.08.006
猜你喜欢
肝博士(2022年3期)2022-06-30 02:48:48
智能建筑电气技术(2022年2期)2022-02-06 02:30:46
海外星云(2021年9期)2021-10-14 07:26:10
商用汽车(2021年4期)2021-10-13 07:16:02
数学物理学报(2020年6期)2021-01-14 01:00:14
数学小灵通(1-2年级)(2020年9期)2020-10-27 03:24:46
作文大王·低年级(2017年11期)2017-12-05 00:08:45
小学生学习指导(低年级)(2017年12期)2017-11-22 06:22:39
中学生数理化·中考版(2017年12期)2017-04-18 12:55:03
青苹果·教育研究版(2016年9期)2016-12-23 11:52:36
