
一种SUMO-Heparinase I融合酶的可溶性表达与纯化
2016-09-07 08:37:04赵善成王振程咏梅陈敬华江南大学药学院江苏无锡2422江南大学糖化学与生物技术教育部重点实验室江苏无锡2422
食品与生物技术学报 2016年3期
赵善成,王振,程咏梅,陈敬华*,2(.江南大学药学院,江苏无锡2422;2.江南大学糖化学与生物技术教育部重点实验室,江苏无锡2422)
一种SUMO-Heparinase I融合酶的可溶性表达与纯化
赵善成1,王振1,程咏梅1,陈敬华*1,2
(1.江南大学药学院,江苏无锡214122;2.江南大学糖化学与生物技术教育部重点实验室,江苏无锡214122)
克隆自肝素黄杆菌(Flavobacteriumheparinum)的肝素酶I(Heparinase I,EC4.2.2.7)广泛应用于低相对分子质量肝素(LMWH,low molecular weight heparin)制备研究。然而,该酶在大肠杆菌表达体系中易形成包涵体,限制了其大量应用。将可溶性配体SUMO(small ubiquitin-like modifier,小泛素类似修饰蛋白质)与C端带有6×His标签的肝素酶基因片段N-端融合表达,菌体总蛋白质和可溶性蛋白质SDS-PAGE电泳表明,N端融合SUMO的肝素酶I可溶性表达比率显著提高;通过镍-亲和柱层析得到均一纯化的融合肝素酶I,融合酶酶学性质表明该融合酶可直接应用于肝素降解而无需切除SUMO标签,为肝素酶I的广泛应用提供了良好的技术基础。关键词:肝素酶I;小泛素类似修饰蛋白质;融合表达;可溶性表达
肝素酶是一类特异性降解肝素和类肝素多糖的裂解酶,可用于病人体外治疗血液中残存肝素的有效去除、血液肝素浓度水平的测定、肝素类多糖的序列分析,以及真核细胞表面肝素类多糖的结构-功能研究[1-3]。其中,肝素酶I降解肝素制备低相对分子质量肝素以质量高、条件温和、易控制和环境友好等优点[4],受到了研究人员越来越多的关注。
相较于肝素黄杆菌发酵法制备肝素酶I,采用pET表达系统在大肠杆菌中重组表达肝素酶I,可显著提高肝素酶I的产量和产品纯度[5]。然而,大肠杆菌表达体系中肝素酶I的非可溶性表达和易失活极大地限制了其大规模应用。近年来发展起来的融合表达策略采用基因工程手段将编码靶蛋白质的基因片段与编码可溶性配体的基因片段融合共表达,可以降低重组蛋白质的降解,改善蛋白质折叠,是一种有效提高蛋白质可溶性从而促进其蛋白质表达水平的方法[6]。之前的研究发现,选用MBP (maltose-bindingprotein)、GST(glutathioneS-transferas)这类相对分子质量较大的标签融合表达肝素酶I可显著改善其可溶性[7-8],但是需要在较低的诱导温度如15℃以下表达靶蛋白质,不利于大规模工业化生产,而CBD(cellulose-binding domain)融合的肝素酶I表达后依然主要以包涵体形式存在[9]。除了这些较大的标签蛋白质之外,组氨酸等较小相对分子质量标签的融合酶易于纯化却无助于改善可溶性。SUMO(small ubiquitin-like modifier)是一种相对分子质量只有11.5 kDa的较小的标签蛋白质[10],已成功应用于严重急性呼吸综合征冠状病毒3CL蛋白酶、核衣壳蛋白质、膜蛋白质以及基质金属蛋白酶13(matrixmetalloprotease-13,MMP13)、生长分化因子8(growth differentiating factor-8,GDF8)的可溶性表达[11-12]。除此之外,去除SUMO标签所用的SUMO蛋白酶具有高保真性,可精确识别SUMO标签的三级结构,因此剪切时会保留靶蛋白质的末端序列尤其是N-端与生物活性密切相关的序列[13]。
基于上述考虑,拟选用SUMO这一相对分子质量更小的助溶性配体标签,将SUMO标签蛋白基因片段与肝素酶I基因片段通过重叠PCR方法构建重组pET-30a-SUMO-Hep I表达载体,将SUMO蛋白质与肝素酶I N-端融合表达,探索其对于肝素酶I可溶性表达的促进作用。
1 材料与方法
1.1材料和仪器
1.1.1菌种、质粒和引物E.coli DH5α,E.coli BL21 (DE3),E.ColiRosseta(DE3),pMD-19T-SUMO,pMD-19T-Hep I和pET-30a(+),pET-30a(+)-Hep I质粒,作者所在实验室保存;Ulp1蛋白酶,作者所在实验室制备并保存;pMD-19T(simple),购自大连宝生物工程有限公司;PCR引物F1:5′-CATATGTCGGACTCAGAAGTCAATCAA-3′(Nde I),R1:5′-CCGGATTTTTTTTGCTGACCTCCAATCTG TTCG-3′,F2:5′-GATTGGAGGTCAGCAAAAAAAAT CCG-3′,R2:5′-GGATCCGATCTGGCAGTTCGCTG-3′(BamH I),均由上海生物工程有限公司合成。
1.1.2工具酶、试剂盒和仪器Ex Taq酶,T4 DNA连接酶,限制性内切酶Nde I和BamH I,购自大连宝生物工程有限公司;PCR产物回收试剂盒和质粒小量提取试剂盒,购自上海生物工程有限公司;蛋白质纯化系统(GE AKTA prime plus),HisTrap HP,购自美国GE公司。
1.2方法
1.2.1SUMO-Hep I基因扩增和重组表达质粒pET-30a(+)-SUMO-Hep I的构建采用重叠PCR方法扩增SUMO-HepI,扩增过程见图1。PCR反应条件为:98℃变性10 s,55℃退火30 s,72℃延伸1 min,共30个循环;72℃充分延伸10 min。PCR产物经质量分数2%琼脂糖凝胶电泳分离,PCR产物回收试剂盒回收后与pMD-19T(simple)克隆载体16℃连接过夜。连接产物pMD-SUMO-Hep I转化E. coli DH5α,通过Amp抗性和蓝白斑筛选出阳性转化子。对经测序验证的重组克隆载体pMD-SUMOHep I和表达载体pET-30a(+)进行双酶切并回收目的片段,并在T4 DNA连接酶作用下16℃连接过夜,连接产物命名为pET-30a(+)-SUMO-Hep I。pET-30a(+)-SUMO-Hep I转化大肠杆菌E.coli Rosseta (DE3),从LB平板挑选单克隆,接种于10 mL LB培养基(含卡那霉素50 μg/mL)中,37℃220 r/min过夜培养,甘油终质量分数15%保种并送上海生工测序验证。重组表达质粒pET-30a(+)-SUMO-Hep I构建流程见图2。
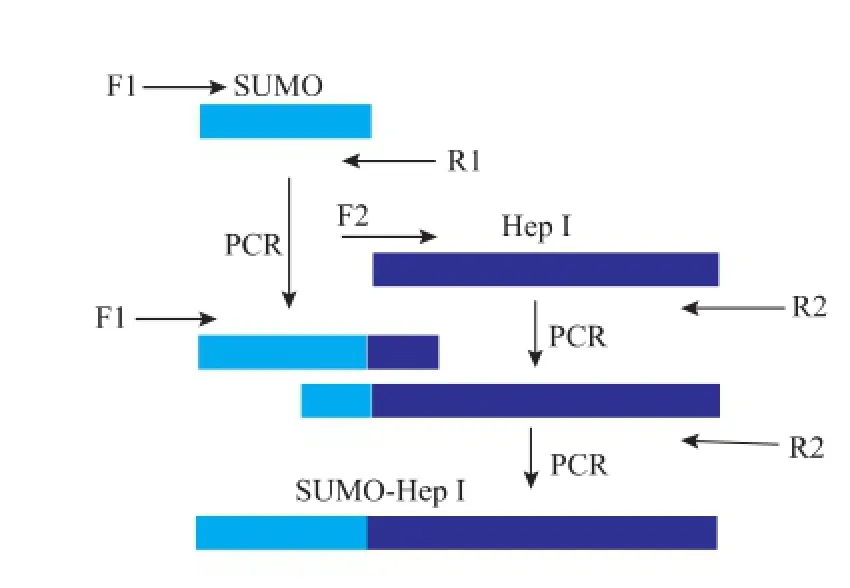
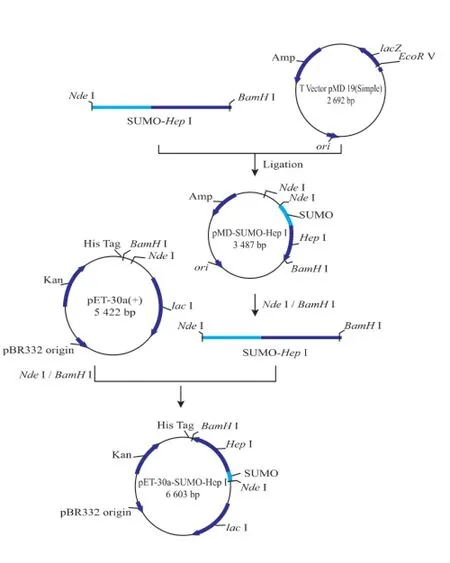
图2 重组表达质粒pET-30a(+)-SUMO-Hep I的构建示意Fig.2 Diagram of construction of recombinant expression plasmid pET-30a(+)-SUMO-Hep I
1.2.2Hep I-6×His和SUMO-HepI-6×His重组蛋白质的表达将经测序验证的保种pET-30a(+)-SUMO-HepI大肠杆菌E.coli Rosseta(DE3),以体积分数1%接种于两瓶10 mL的LB培养基(含卡那霉素50 μg/mL),37℃220 r/min过夜培养,作为种子液。再以体积分数1%的接种量接种到50 mL LB培养基(含卡那霉素50 μg/mL),37℃220 r/min培养,当OD600达0.6~0.8时,加终浓度为0.2 mmol/L的IPTG,25℃220 r/min诱导表达10 h。pET-30a (+)-Hep I表达菌株E.coli BL21(DE3)在活化后亦按照上述条件诱导表达。
1.2.3Hep I-6×His和SUMO-Hep I-6×His重组蛋白质的纯化4℃、9 000 r/min离心10 min收集菌体,Binding Buffer(20 mmol/L Tris,0.5 mol/L NaCl,10 mmol/L咪唑,pH 7.4)重悬,超声破碎(200 W,超声5 s,间歇8 s,工作99个循环,共3次)后12 000 r/ min离心30 min,上清液用0.45 μm水系膜过滤,过滤液用蛋白质纯化系统GE AKTA prime plus进行纯化。HisTrap HP(5 mL)亲和层析纯化条件:采用阶段梯度洗脱模式,先用5个柱体积质量分数4%的ElutionBuffer(20mmol/LTris,0.5mol/LNaCl,500mmol/L咪唑,pH 7.4)冲洗结合力的杂蛋白质,再采用体积分数50%的Elution Buffer洗脱目的蛋白质,体积流量5 mL/min收集洗脱液。纯化完毕,BCA法分别测定Hep I-6×His、SUMO-Hep I-6×His的产量。将纯化的SUMO-Hep I-6×His与Ulp 1蛋白酶按照质量比1 000∶1混合均匀后置于4℃酶切1 h,并按照上述条件进行镍柱亲和纯化。
1.2.4SUMO-Hep I-6×His酶活表征参照文献[14]方法对SUMO-Hep I-6×His的相对酶活进行测定:在200 μL pH 7.0的缓冲液(0.05 mol/L醋酸钠、5 mmol/ L醋酸钙)中加入1 mg肝素,再加入20 μL保存于质量分数0.1%BSA中的HepI。30℃水浴反应10min,立即加入0.06 mol/L盐酸2 mL终止反应。3 000 r/ min离心10 min,取上清液测定232 nm处吸光值。一个酶活力单位是指30℃pH 7.0的条件下,在1 min内产生1 μmol具有Δ4,5不饱和糖醛所需要的酶量,酶活(IU/mL)计算
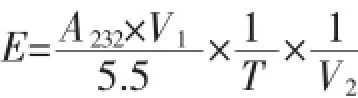
上式中,A232为在波长232 nm下测定的紫外吸收值;V1为反应液总体积(mL);V2为加入酶的体积(mL);T为反应时间(min);反应系数为5.5(μmol/mL)。
测定重组融合蛋白质Hep I-6×His和SUMOHep I-6×His的各纯化步骤中蛋白质含量和酶活性。
2 结果与讨论
2.1SUMO-Hep I编码序列的扩增
见图3,PCR扩增产物经2 g/dL琼脂糖凝胶电泳后,在约300、1 100、1 400 bp处可清楚地观察到DNA条带,大小和SUMO编码片段、Hep I编码片段,以及SUMO-Hep I编码片段理论值291、1 155、1 446 bp一致。
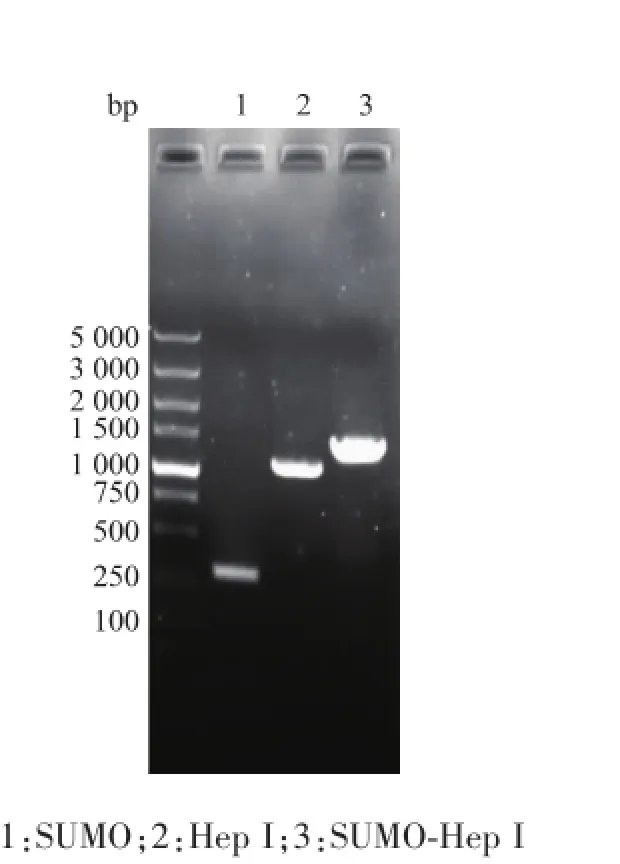
图3 基因电泳图Fig.3 Gene electrophoresis
2.2pET-30a(+)-SUMO-Hep I重组质粒测序鉴定结果
表达质粒pET-30a(+)-SUMO-Hep I送至上海华大基因公司测序,测序结果显示重组表达质粒与设计基因序列一致,表明重组表达质粒构建成功。
2.3Hep I-6×His和SUMO-Hep I-6×His重组蛋白质的表达
如图4所示,在诱导温度25℃条件下,融合SUMO标签的肝素酶I呈高水平表达,且融合后可溶性组分占总蛋白质组分由融合SUMO标签前的质量分数(24.28±1.63)%显著提高至(94.52±3.99)%(图5),证明采用SUMO融合表达肝素酶I是一种可提高靶蛋白质可溶性表达的有效策略。
2.4Hep I-6×His和SUMO-Hep I-6×His重组蛋白质的纯化
如图6所示,SDS-PAGE电泳结果表明,在43.8 kDa、63.8 kDa分别出现Hep I-6×His、SUMO-Hep I-6×His预期的单一蛋白质条带。BCA法测定纯化酶的产量为Hep I-6×His和SUMO-Hep I-6×His分别为2.2 mg/(0.3 g干质量菌体)和3.4 mg/(0.3 g干质量菌体),进一步表明SUMO标签的融合确有助于提高融合酶的可溶性从而提高其表达水平。
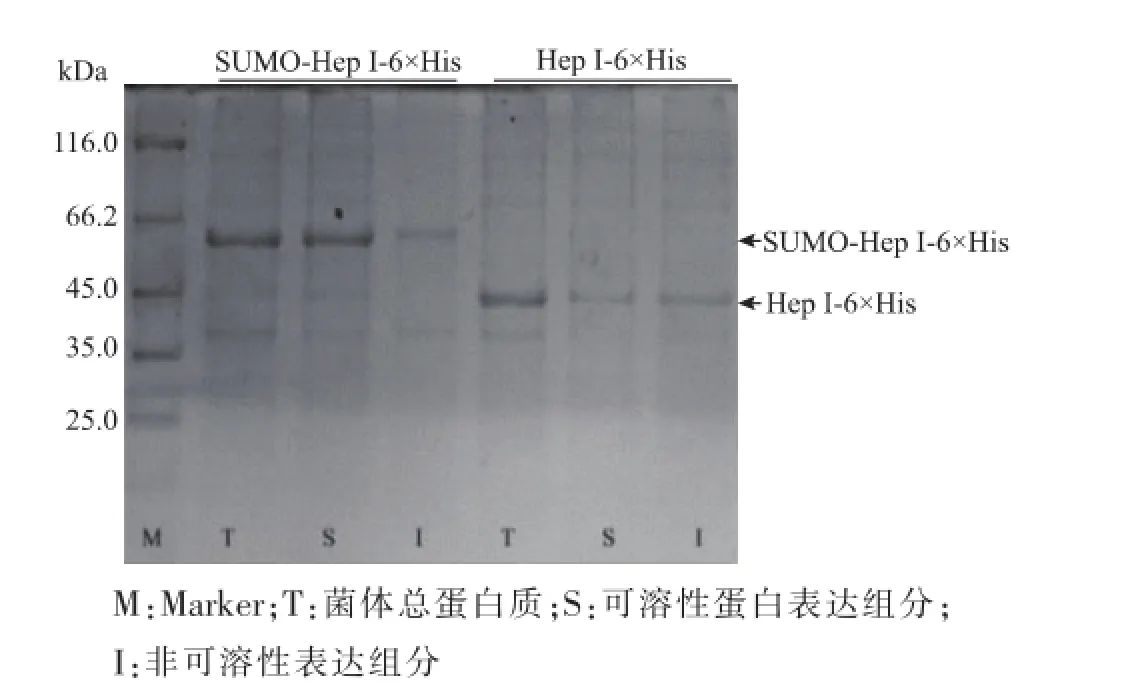
图4 SUMO-Hep I-6×His融合表达的SDS-PAGE分析Fig.4 SDS-PAGE analysis of fusion expression of SUMOHep I-6×His
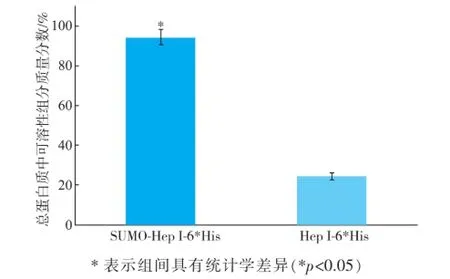
图5 SUMO-Hep I-6×His表达的可溶性组分与Hep I-6× His表达的可溶性组分的比较Fig.5 Comparison of the portion of soluble expression of SUMO-Hep I-6×His with Hep I-6×His

图6 重组SUMO-Hep I-6×His融合蛋白质纯化效果的SDS-PAGE鉴定Fig.6 SDS-PAGE identification of purification efficiency of recombinant SUMO-Hep I-6×His
2.5SUMO-Hep I-6×His酶活表征
重组融合蛋白质SUMO-Hep I-6×His的各纯化步骤中蛋白质含量和酶活测定结果如表1所示。

表10 .3 g干质量菌体内蛋白质纯化和酶活测定结果Table 1 Protein purification and enzymatic activity in 0.3 g dry cell mass
结果表明,使用SUMO标签融合不仅可提高肝素酶I可溶性表达水平、总酶活和比酶活,每0.3 g干质量菌体总的可溶性含粗酶的蛋白质由178.7 mg提高到227.1 mg,而且可提高其镍柱亲和纯化的回收率和纯化倍数,回收率由64.2%提高到79.2%,证明SUMO标签在改善肝素酶I镍柱亲和纯化效果方面具有一定促进作用。
此外,SUMO-Hep I-6×His的最适pH和最适温度表征结果(图7)表明,SUMO标签在提高肝素酶I表达的可溶性的同时,融合肝素酶I的最适pH和最适温度未发生改变,与天然肝素酶I基本一致,仍为pH 7.0和30℃[15]。因此SUMO-Hep I-6×His可无需切除SUMO标签直接使用,方便快捷。切除SUMO标签后,单位物质的量比酶活恢复到融合之前,表明SUMO标签有利于肝素酶I酶活的提升,其机制或影响到肝素酶I三级结构而改变其底物亲和力或与其催化效率有关,这有待进一步深入研究。
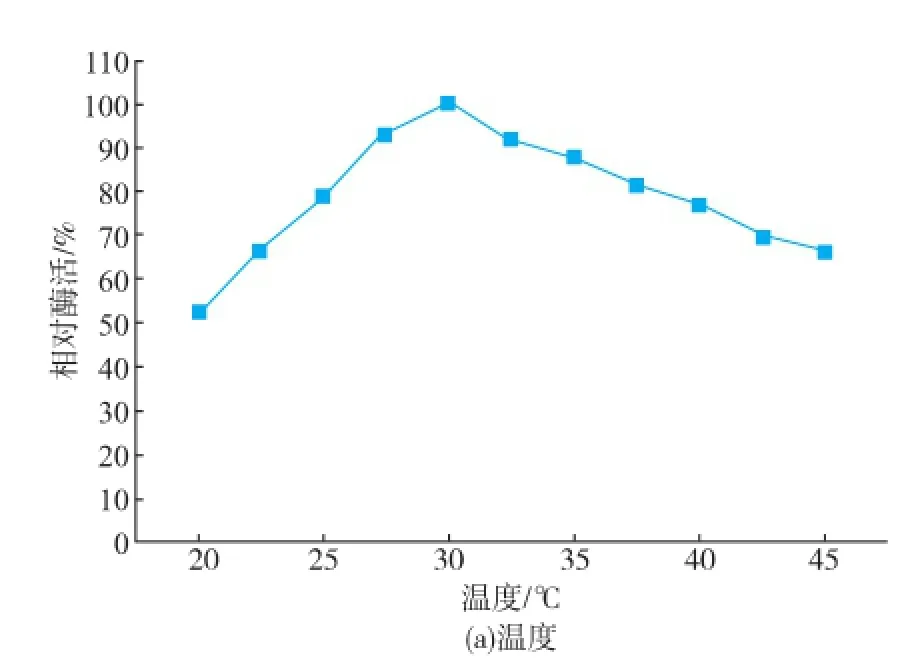
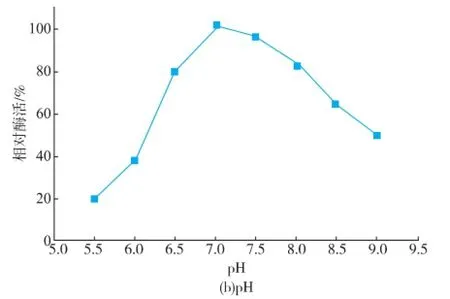
图7 SUMO-Hep I-6×His的最适温度和最适pHFig.7 Optimum temperature and pH of SUMO-Hep I-6× His
3 结语
难以经济高效地表达肝素酶I,一直是限制其大规模工业应用尤其是在酶法制备低相对分子质量肝素领域应用的瓶颈之一。使用SUMO这种较小相对分子质量的标签配体蛋白质,融合表达蛋白质,可在有效提高靶蛋白质可溶性的同时,避免空间位阻效应对靶蛋白质活性的影响。本研究中利用该性质成功构建了一种SUMO-Hep I-6×His融合酶,提高了肝素酶I的可溶性表达,肝素酶I的产量由2.2 mg/(0.3 g干质量菌体)提高到3.4 mg/(0.3 g干质量菌体),可溶性表达比率由(24.28±1.63)%显著提高至(94.52±3.99)%,且无需切除标签可直接应用于肝素降解,为肝素酶I的广泛应用提供了一条成功的工艺路线。
[1]Ernst S,Venkataraman G,Winkler S,et al.Expression in Escherichia coli,purification and characterization of heparinaseI from Flavobacteriumheparinum[J].Biochem J,1996,15(315):589-597.
[2]刘卫超,程咏梅,邓超,等.GST-His-HeparinaseⅠ双标签融合蛋白的原核表达与纯化[J].工业微生物,2014,44(5):51-56. LIU Weichao,CHENG Yongmei,DENG Chao,et al.Prokaryotic expression and purification of GST-His-Heparinase I doublelabelled fusion protein[J].Industrial Microbiology,2014,44(5):51-56.(in Chinese)
[3]Linhardt R J,Turnbull J E,Wang H M,et al.Examination of the substrate specificity of heparin andheparan sulfatelyases[J]. Biochemistry,1990,29:2611-2617.
[4]Ye Fengchun,Kuang Ying,Chen Shuo,et al.Characteristics of low molecular weight heparin production by an ultrafiltrationmembrane bioreactor using maltose binding protein fused heparinase I[J].Biochemical Engineering Journal,2009,46:193-198.
[5]Weitz J I.Low-molecular-weight heparins[J].N Engl J Med,1997,337:688-698.
[6]Davis G D,Elisee C,Newham D M,et al.New fusion protein systemsdesigned to give soluble expression in Escherichia coli[J]. Biotechnol Bioeng,1999,65:382-388.
[7]Chen Yin,Xing Xinhui,Lou Kai.Construction of recombinant escherichia coli for over-production of soluble heparinase I by fusion to maltose-binding protein[J].Biochem Eng J,2005,23:155-159.
[8]Luo Yongde,Huang Xingqiang,Mc Keehan W L.High yield,purity and activity of soluble recombinant bacteroides thetaiotaomicron GST-heparinase I from Escherichia coli,Arch[J].Biochem Biophys,2007,460:17-24.
[9]Shpigel E,Goldlust A,Efroni G,et al.Immobilization of recombinant heparinase I fused to cellulose-binding domain[J]. Biotechnol Bioeng,1999,65:17-23.
[10]Tatham M H,Jaffray E,Vaughan O A,et al.Polymeric chains of SUMO-2 andSUMO-3 are conjugated to protein substrates by SAE1/SAE2 andUbc9[J].J Biol Chem,2001,276:35368-35374.
[11]Zuo Xun,Mattern M R,Tan R,et al.Expression andpurification of SARS Coronavirus proteins using SUMO fusions[J].Protein Express Purif,2005,42:100-110.
[12]Marblestone J G,Edavettal S C,Lim Y,et al.Comparison of SUMO fusion technology with traditional gene fusion systems:enhancedexpression and solubility with SUMO[J].Protein Sci,2006,15:182-189.
[13]Malakhov M P,Mattern M R,Malakhova O A,et al.SUMO fusions and SUMO-specific proteasefor efficient expression and purification of proteins[J].J Struct Funct Genomics,2004(5):75-86.
[14]Yu Ping,Wu Yan.Expression of the heparinase gene from Flavobacteriumheparinum in Escherichia coli and its enzymatic properties[J].See comment in PubMed Commons Below Carbohydr Polym,2012,90(1):348-352.
[15]Yang Victor C,Robert J Linhardt,Howard Bernstein,et al.Purification and characterization of heparinase from Flavobacterium heparinurn[J].J B C,1985,260(3):1849-1857.
Soluble Expression and Purification of the SUMO-Heparanase I Fusion Enzyme
ZHAO Shancheng1,WANG Zhen1,CHENG Yongmei1,CHEN Jinghua*1,2
(1.School of Pharmaceutical Science,JiangnanUniversity,Wuxi 214122,China;2.Key Laboratory of Carbohydrate Chemistry and Biotechnology Ministry of Education,Jiangnan University,Wuxi 214122,China)
Heparinase I cloned from Flavobacterium heparinum was widely used in the preparation of LMWH(low molecular weight heparin).However,the formation of inclusion bodies when expressed in Escherichia coli limits its application in a large scale.In this study,the soluble partner,small ubiquitin-like modifier(SUMO),was fused to the N-terminus of Hep I gene with a C-terminal 6×histidine tag.SDS-PAGE analysis of total protein in bacteria and soluble fractions indicated that the portion of the soluble Heparinase I fused with SUMO to the N-terminus was significantly enhanced.After purified by the nickel-chelate chromatography,the resulting fusion enzyme could be used directly without removing the fusion SUMO tag,which made the wide application of Heparinase I feasible.
Heparinase I,small ubiquitin-like modifier,fusion expression,soluble expression
O 629.12
A
1673—1689(2016)03—0318—06
2014-12-15
教育部博士点基金项目(20110093110008)。
赵善成(1986—),男,安徽六安人,工学博士,主要从事肝素类多糖和相关酶的研究。E-mail:zhaoshancheng2008@163.com
陈敬华(1971—),男,湖北黄石人,理学博士,教授,博士研究生导师,主要从事生物大分子和生物功能材料的研究。
E-mail:jhchenwhut@126.com
