
肝X受体激动剂上调人肾小球内皮细胞血栓调节蛋白表达的机制和作用*
2016-08-08 02:58丁涵露李怡冯韵霖陈瑾钟翔王楠汪伟张萍王莉
中国现代医学杂志 2016年5期
丁涵露,李怡,冯韵霖,陈瑾,钟翔,王楠,汪伟,张萍,王莉
[1.四川省医学科学院(四川省人民医院)肾内科,四川 成都 610072;2.四川省成都市第二人民医院肾内科,四川 成都 610017]
·论著·
肝X受体激动剂上调人肾小球内皮细胞血栓调节蛋白表达的机制和作用*
丁涵露1,李怡1,冯韵霖1,陈瑾1,钟翔1,王楠2,汪伟1,张萍1,王莉1
[1.四川省医学科学院(四川省人民医院)肾内科,四川 成都 610072;2.四川省成都市第二人民医院肾内科,四川 成都 610017]
目的探讨肝X受体(LX R)激动剂T0901317上调人肾小球内皮细胞(H R G EC)血栓调节蛋白(TM)表达的机制和作用。方法W es t ern bl ot检测25 m m ol高糖和2μm ol T0901317刺激后的H R G EC上IκBα、磷酸化IκBα、核转录因子-κB(N F-κB)p65、磷酸化N F-κB p65表达;免疫共沉淀法检测LX R与P300之间有无结合;重组腺病毒AdTM shR N A转染H R G EC,观察其对高糖下H R G EC分泌炎症介质的影响。结果T0901317能显著降低高糖刺激后的IκBα和N F-κB p65磷酸化(P<0.05),LX R-α沉默后N F-κB活性增强;2μm ol T0901317刺激H R G EC 24 h后以C o-IP检测LX R与P300的表达上调;T0901317明显抑制高糖刺激下的H R G EC培养上清中TN F-α、IL-1β浓度(P<0.05),AdTM s hR N A转染H R G EC后,有或无T0901317刺激,H R G EC培养上清中TN F-α、IL-1β浓度与高糖组比较,差异无统计学意义(P>0.05)。结论LX R可能通过与P300发生相互作用阻断了N F-κB与P300之间的竞争性结合而提高TM的表达并抑制炎症介质的分泌。
人肾小球内皮细胞;肝X受体;血栓调节蛋白;核转录因子-κB
人肾小球内皮细胞(hum an renal gl om erul ar endot hel i al cel l s,H RGEC)炎性损伤是慢性肾脏病尿蛋白形成的重要原因,既往研究多集中于致炎机制,而对H RGEC内源性抗炎机制的关注很少。血栓调节蛋白(t hrom bom odul i n,TM)通过促进活化蛋白C形成及抑制高迁移率族蛋白1等途径发挥重要的内源性抗炎功能[1]。肝X受体(l i ver X recept or,LXR)是核受体家族成员中一种重要的核受体,T0901317是LXR的高亲和力人工合成配体,通常用于实验研究。在哺乳动物中存在LXRα和LXRβ两种亚型,LXR激动剂与LXR结合后激活靶基因的转录而参与胆固醇、脂类和葡萄糖等物质代谢[1-2],以及发挥重要的抗炎作用[3-4]。前期的研究发现培养的H RGEC表达LXRα、LXRβ,LXR激动剂主要通过LXRα上调TM表达,但LXR激动剂上调H RGEC TM表达的机制不明,以荧光素酶报告实验发现TM启动子区域(-2494~+160)无LXR结合点。核转录因子-κB (nucl ear t ranscri pt i on f act or-κB,NF-κB)需要与协同转录辅活化因子P300结合才能发挥转录活性[5-6],有人发现,核受体家族成员PPARγ依赖配体激活后,通过与NF-κB竞争结合P300而起到抑制核转录因子-κB信号通路活性的作用[7],同样作为核受体家族的全反式维甲酸也是通过竞争性结合P300而抑制肿瘤坏死因子-α(t um or necrosi s f act or-α,TNF-α)诱导的TM表达下降[5,8],因此笔者推测LXR可能通过竞争性结合P300而抑制NF-κB活性,故本研究拟检测高糖和LXR激动剂T0901317刺激后的H RGEC上NF-κB信号通路活性改变,在此基础上免疫共沉淀法检测LXR与P300之间有无结合,最后以重组腺病毒AdTM shRNA转染H RGEC,观察其对高糖刺激下H R GEC分泌炎症介质的影响,以探讨LXR激动剂上调TM表达的机制和作用。
1 材料与方法
1.1实验试剂
H RGEC和T0901317(美国Si gm a公司),小鼠抗人IκBα抗体、小鼠抗人磷酸化IκBα抗体、小鼠抗人NF-κB p65抗体,小鼠抗人磷酸化NF-κB p65抗体(美国Cel l Si gnal i ng Technol ogy公司)、兔抗人P300抗体、兔抗人TM抗体(美国Sant a公司),小鼠抗人β-act i n抗体、羊抗小鼠IgG、羊抗兔IgG(北京博奥森公司),shRNA-LXRα设计与合成由美国Invi t rogen Li f e Technol ogi es公司完成,白介素-1β(i nt erl euki n-1β,IL-1β)和TNF-α酶联免疫吸 附 法 (enzym e-l i nkedi m m unosorbent assay,ELISA)试剂盒(美国Pi erce公司)。
1.2W est ern bl ot检测LXR激动剂及shR N ALXR α对N F-kB信号通路蛋白的作用
1.2.1实验分组①二甲基亚砜(di m et hyl sul f oxi de,DM SO)对照组;②25 m m ol/L葡萄糖组(Gs组);③25 m m ol/L葡萄糖+2μm ol/L T0901317(Gs+T09组);④25 m m ol/L甘露醇组(M a组);⑤shRNALXRα+25 m m ol/L葡萄糖+2μm ol/L T0901317;⑥shRNA-C ont rol+25m m ol/L葡 萄 糖+2μm ol/L T0901317,其中⑤⑥组分别给予含shRNA-LXRα或shRNA-Cont rol病毒液转染,24 h更换成完全培养基继续培养至48 h,将病毒感染48 h后的细胞分别予以葡萄糖和/或T0901317处理24 h。
1.2.2实验方法上述各实验分组在药物处理24 h后收集细胞加蛋白裂解液提取总蛋白,检测细胞总蛋白浓度后取50μg蛋白样品经十二烷基硫酸钠-聚丙烯酰胺(sodi um dodecyl sul f at e-pol yacryl am i de gel el ect rophoresi s,SDS-PAGE)凝胶电泳后,13 V恒压过夜,将凝胶中蛋白质转移至聚偏二氟乙烯(pol yvi nyl i dene f l uori de,PVDF)膜上。PVDF膜以含5%脱脂奶粉的TBST室温封闭2 h,分别加入phosphor-NF-κB p65(1∶1 000)、NF-κB p65(1∶1000)、phosphor-IκBα(1∶1 000)、IκBα(1∶1 000)和β-act i n(1∶1 000)单克隆抗体室温反应2 h,山羊抗兔、山羊抗小鼠IgG二抗(1∶1 000)室温孵育2 h,化学发光显色,每组实验均重复3次,用Gel Pro Anal yzer 3.2图像分析软件分析条带灰度值,取平均值做为实验结果。
1.3免疫共沉淀法检测LXR与P300之间有无结合
分别将有或无2μm ol/L T0901317处理24 h的H RGEC做LXR与P300的免疫沉淀反应,用含1% Tri t on X-100磷酸盐缓冲溶液(phosphat e buf f er sal i ne,PBS)(含蛋白酶抑制剂和磷酸酶抑制剂)提取细胞匀浆,4℃、14000×g离心30m i n后,将裂解液分成4等份,一份加入10μl兔IgG抗体作为阴性对照组,一份不加抗体作为空白对照组,一份加入0.2μg P300抗体,一份加入0.2μg TM抗体,上述各组分别在4℃孵育过夜,然后与100μl蛋白A/G琼脂糖珠子摇床4℃孵育6 h,2 000 r/mi n离心2 m i n,去上清液,以1.5 m l预冷的PBS反复离心洗涤珠子5次,去上清液,以5×上样缓冲液20μl于100℃变性10 m i n,2 000 r/mi n离心5 m i n,收集上清液,置于-20℃冰箱保存,用于10%SDS-PAGE电泳。
1.4ELI SA观察腺病毒AdTM shR N A对高糖刺激下H R G EC分泌炎症介质的影响
将构建成功的TM小分子干扰RNA的腺病毒AdTM shRNA用于以下实验。
1.4.1实验分组①25 m m ol/L葡萄糖组(Gs组);②25 m m ol/L葡萄糖+2μm ol/L T0901317(Gs+T09组);③25 m m ol/L葡萄糖+AdTM shRNA+2μm ol/L T0901317(Gs+AdTM shRNA+T09组);④25 m m ol/L葡萄糖+Adcont rol+2μm ol/L T0901317(Gs+AdCt rl+ T09组);⑤25 m m ol/L葡萄糖+AdTM shRNA+(Gs+ AdTM shRNA组);⑥25 m m ol/L葡萄糖+Adcont rol (Gs+AdCt rl组),其中③~⑥组分别给予含AdTM shRNA或Adcont rol病毒液转染,24 h更换成完全培养基继续培养至48 h,将病毒感染48 h后的细胞分别予以葡萄糖和/或T0901317处理24 h。
1.4.2实验方法上述各实验分组药物作用24 h后收集上清液,以ELISA检测细胞培养上清液中TNF-α、IL-1β浓度。
1.5统计学方法
采用SPSS 13.0统计软件进行数据分析,实验数据以均数±标准差(±s)表示,多组数据平均值比较用单因素方差分析(ANOVA),组间比较用Bonf erroni分析进行,两组间比较用t检验,所有检验均为双侧检验,P<0.05为差异有统计学意义。
2 结果
2.1W est ern bl ot检测LXR激动剂抑制N F-κB信号通路活性
Gs组与对照组的IκBα、NF-κB p65水平比较,经t检验,差异有统计学意义(tI κBα=-20.684,PI κBα= 0.000,tNF-κB p65=-13.133,PNF-κB p65=0.000),表明高糖刺激后,IκBα磷酸化增加和p-p65(核/浆)水平升高(见表1)。Gs+T09组与Gs组的p-IκBα、p-p65(核/浆)水平比较,经t检验,差异有统计学意义(tp-I κBα= -4.867,Pp-I κBα=0.008,tp-p65=-10.384,Pp-p65=0.000),Gs+T09组的p-IκBα、p-p65(核/浆)水平较Gs组降低,表明T0901317能降低高糖刺激后的p-IκBα、p-p65(核/浆)水平(见图1A、1B和表1)。shRNA-LXRα组与对照组的p-IκBα、p-p65(核/浆)水平比较,经t检验,差异有统计学意义(P<0.05),shRNA-LXRa 组IκBα磷酸化增加,p-p65(核/浆)水平升高,表明shRNA-LXRα干扰后IκBα磷酸化增加,p-p65(核/浆)水平升高(见图1C、1D和表2)。
2.2免疫共沉淀法检测LXR与p300之间有结合
2μm ol T0901317刺激H RGEC 24 h后用Co-IP检测发现LXR与P300的表达上调,在以LXR抗体免疫共沉淀下来的蛋白复合物中可检测到P300蛋白,在以P300抗体免疫共沉淀下来的蛋白复合物中可检测到LXR蛋白(见图2)。由此可推断出T0901317可以促进LXR与P300之间的结合,证明LXR与P300之间是有相互作用的。由于NF-κB需要与P300结合才能发挥转录活性,因此结合本实验发现T0901317抑制NF-κB信号通路蛋白活性,笔者推测LXR可能竞争性与P300结合,阻断了NF-κB与P300之间的结合,NF-κB活性下降从而TM表达提高。
2.3AdTM shR N A转染H R G EC后抑制高糖下H R G EC分泌的炎症介质
Gs+T09组与高糖组的TNF-α、IL-1β水平比较,经t检验,差异有统计学意义(tTNF-α=15.728,PTNF-α= 0.000,tI L-1β=13.090,PI L-1β=0.000),Gs+T09组的TNF-α、IL-1β水平较高糖组降低,表明T0901317能降低高糖刺激后的 TNF-α、IL-1β 水平。Gs+AdTM shRNA+T09组的TNF-α、IL-1β水平与高糖组比较,经t检验,差异无统计学意义(tTNF-α= 1.880,PTNF-α=0.133,tI L-1β=1.654,PI L-1β=0.173);Gs+ AdTM shRNA组的TNFα、IL-1β水平与高糖组比较,经t检验,差异无统计学意义(tTNF-α=2.76,PTNF-α= 0.051,tI L-1β=-0.533,PI L-1β=0.623), 两 组 的TNF-α、IL-1β 水平较高糖组无明显降低,表明AdTM shRNA转染H RGEC后,无论有或无T0901317刺激,H RGEC培养上清中TNF-α、IL-1β水平都无明显降低,提示TM可能具有减轻高糖刺激下肾小球内皮细胞炎症反应的作用。见表3和图3。
表1 各组p-I kBα光密度和p-p65(核/浆)比较 (n=3,±s)

表1 各组p-I kBα光密度和p-p65(核/浆)比较 (n=3,±s)
组别P值对照组 6 0 . 6 7 ± 2 . 5 1 7 p -I k Bα光密度/ % F 值P 值p -p 6 5(核/浆)F 值8 5 . 6 7 ± 2 . 5 1 7 G s组 1 2 2 . 3 3 ± 4 . 5 0 9 1 3 5 . 0 0 ± 6 . 0 0 0 G s + T 0 9组 7 0 . 6 7 ± 2 . 5 1 6 1 1 4 . 0 0 ± 4 . 0 0 0 M a组 4 7 . 0 0 ± 4 . 0 0 0 6 4 . 0 0 ± 4 . 0 0 0 2 6 5 . 1 8 8 0 . 0 0 0 1 5 7 . 2 3 8 0 . 0 0 0
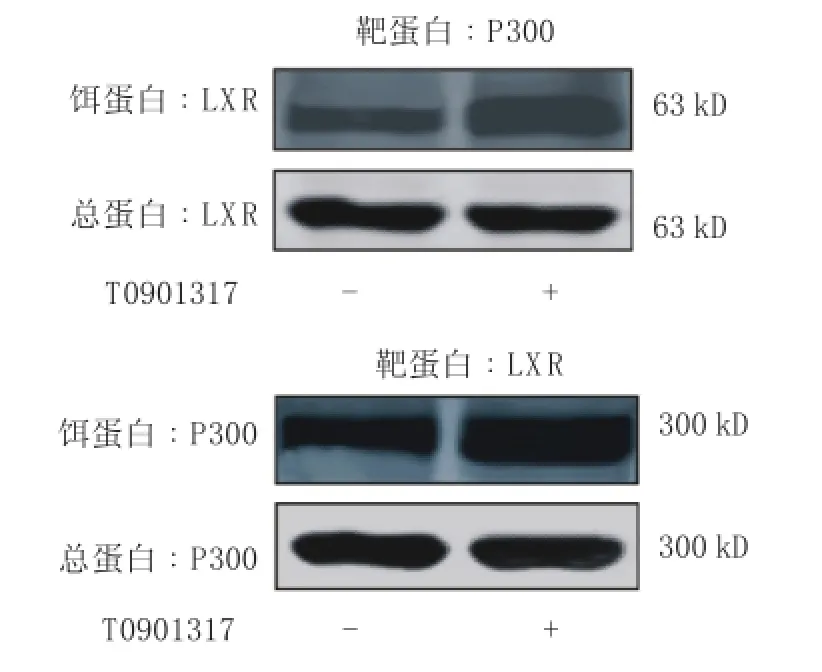
图2 免疫共沉淀法检测LXR与P300之间有结合
表2 shR N A-LXR α干扰后各组p-I kBα光密度和p-p65(核/浆)比较 (%,n=3±s)
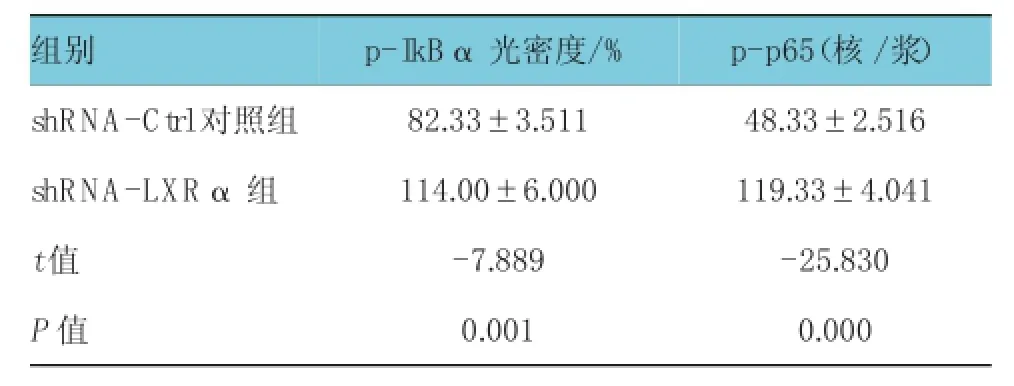
表2 shR N A-LXR α干扰后各组p-I kBα光密度和p-p65(核/浆)比较 (%,n=3±s)
组别p -p 6 5(核/浆)s h R N A -C t r l对照组 8 2 . 3 3 ± 3 . 5 1 1 4 8 . 3 3 ± 2 . 5 1 6 s h R N A -L X Rα组 1 1 4 . 0 0 ± 6 . 0 0 0 1 1 9 . 3 3 ± 4 . 0 4 1 t值 -7 . 8 8 9 -2 5 . 8 3 0 P值 0 . 0 0 1 0 . 0 0 0 p -I k Bα光密度/ %
表3 T0901317抑制高糖刺激下H R G EC分泌TN Fα、I L-1β (n=3,pg/m l,±s)
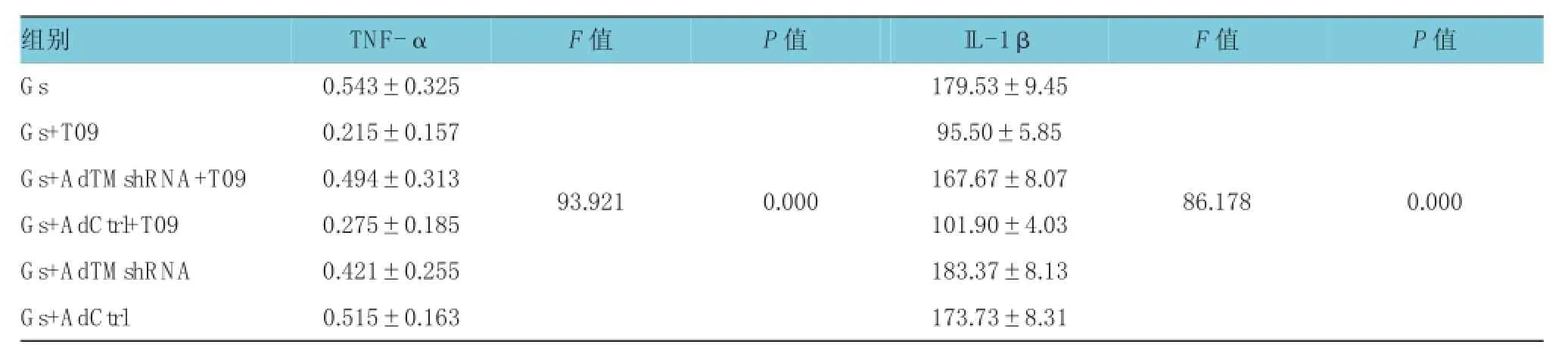
表3 T0901317抑制高糖刺激下H R G EC分泌TN Fα、I L-1β (n=3,pg/m l,±s)
组别P 值G s 0 . 5 4 3 ± 0 . 3 2 5 T N F -α F 值P 值I L -1 β F 值1 7 9 . 5 3 ± 9 . 4 5 G s + T 0 9 0 . 2 1 5 ± 0 . 1 5 7 9 5 . 5 0 ± 5 . 8 5 G s + A d T M s h R N A + T 0 9 0 . 4 9 4 ± 0 . 3 1 3 1 6 7 . 6 7 ± 8 . 0 7 G s + A d C t r l + T 0 9 0 . 2 7 5 ± 0 . 1 8 5 1 0 1 . 9 0 ± 4 . 0 3 G s + A d T M s h R N A 0 . 4 2 1 ± 0 . 2 5 5 1 8 3 . 3 7 ± 8 . 1 3 G s + A d C t r l 0 . 5 1 5 ± 0 . 1 6 3 1 7 3 . 7 3 ± 8 . 3 1 9 3 . 9 2 1 0 . 0 0 0 8 6 . 1 7 8 0 . 0 0 0
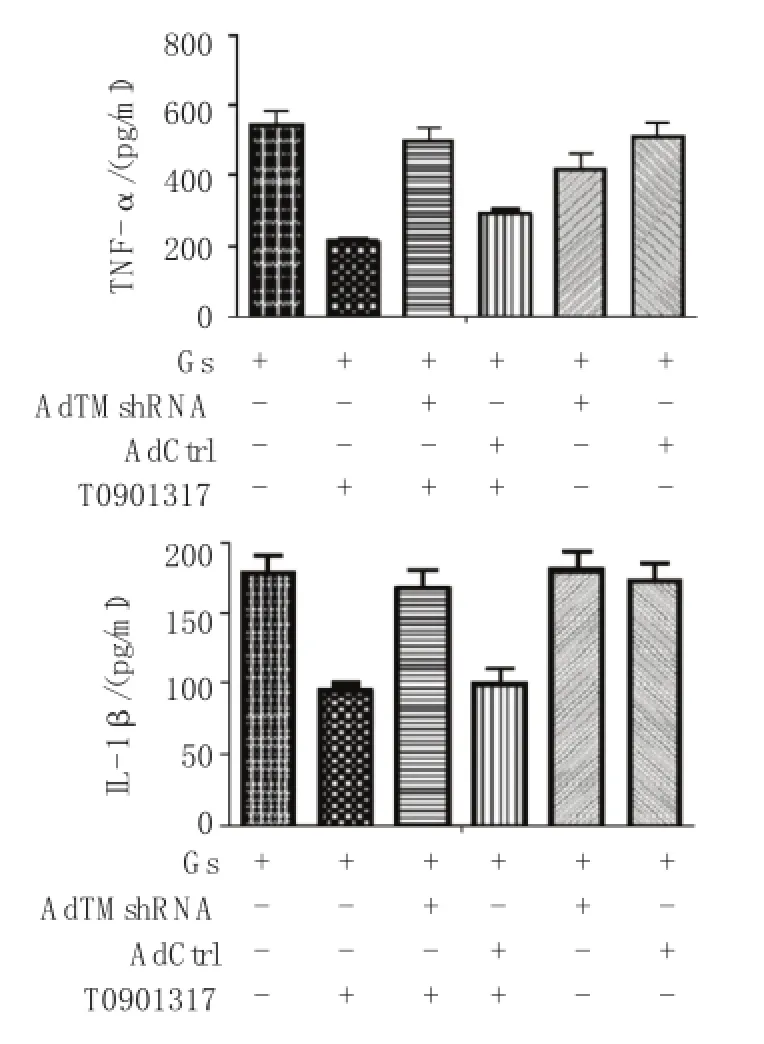
图3 AdTM shR N A转染H R G EC抑制高糖下H R G EC分泌TN F-α、I L-1β
3 讨论
NF-κB是普遍存在于细胞浆中以p50/p65异二聚体为形式的一种快反应转录因子,与NF-κB的抑制性蛋白 IκB(i nhi bi t or,IκB)包括IκBα和IκBβ)结合而呈非活性状态。当细胞受到病原菌等刺激时,IκB激酶因磷酸化而活化并使IκBα发生磷酸化,继而发生泛素化而被26 S蛋白酶体降解。IκBα降解使得p50/p65转位入核,对多种靶基因发挥转录调控作用。p65的核定位信号区的N-氨基端(S276)发生磷酸化是p50/p65异二聚体转位、与靶基因DNA结合及调节靶基因转录所必需的[9-10],已经明确,大多数的炎症介质包括TNF-α、IL-6启动子中包含特异性NF-κB结合保守序列[11-12]。
既往研究报道,NF-κB可通过竞争性结合P300而抑制某些转录因子的活化[13-14],而且LXR启动子区域可能含有NF-κB的结合位点[15-16]。IκBα磷酸化是NF-κB通路得以激活的初始信号,本实验发现高糖刺激后IκBα磷酸化比例明显增加,LXR激动剂T0901317能显著降低高糖刺激后的IκBα磷酸化和NF-κB p65磷酸化。特异性敲低H RGEC上的LXRα,再以高糖刺激及T09013171处理,与无效RNA干扰对照组比较,IκBα磷酸化比例增加(P<0.05),同样磷酸化NF-κB p65水平也显著提高(P<0.01),提示T0901317可抑制NF-κB信号通路蛋白活性,而且这种抑制作用依赖于LXRα的活化,上述实验结果与已有文献报道的LXR激动剂抑制IκB磷酸化和NF-κB p65转位入核而上调缺血心肌细胞血红素加氧酶的表达相一致[16],因此本实验中LXR激动剂促进TM表达有可能与其抑制NF-κB信号通路蛋白的活性有关。
作为核受体家族一员的 LXR活化后抑制NF-κB通路活性的机制虽然尚未完全阐明,但有人发现,核受体家族的另一个重要成员PPARγ能与NF-κB发生生理性相互作用,PPARγ激动剂存在时两者的相互作用增强,并抑制NF-κB驱动炎症因子基因转录[17]。而有人则提出,PPARγ依赖配体激活后,通过与NF-κB竞争结合P300而起到抑制NF-κB信号通路活性的作用[7]。同样作为核受体家族的全反式维甲酸也是通过竞争性结合P300而抑制TNF-α诱导的TM表达下降[5,8],在本实验中,T0901317刺激后的H RGEC上NF-κB信号通路活性下降,加之免疫共沉淀法检测到LXR与P300之间有结合,因此推测LXR活化后,LXR可能通过与NF-κB竞争性结合P300而抑制NF-κB的活性,达到上调TM表达和抑制炎症介质分泌的作用。
LXR-β在多种组织广泛表达,LXR-α主要表达在胆固醇代谢组织[18],之前的研究发现LXR-α、LXR-β表达在H RGEC,LXR-α激活后TM表达增强。本实验发现,转染LXR-α-si RNA后,H RGEC的NF-κB信号通路蛋白活性增加,提示LXR-α活化在H R GEC表达TM中可能起重要作用,鉴于T0901317可同时激活LXR-α、LXR-β[19],因此内源性LXR-β在TM表达中的作用不能完全排除。总之,本实验发现LXR活化可抑制内皮细胞炎症介质分泌,因此靶向LXR-TM的治疗可能会减轻高糖下内皮细胞的炎症反应。
[1]Szant o A,Roszer T.Nucl ear recept ors i n m acrophages:a l i nk bet ween m et abol i sm and i nf l am m at i on[J].FEBS Let t,2008,582(1): 106-116.
[2]Cal ki n AC,Tont onoz P.Li ver X recept or si gnal i ng pat hways and at heroscl erosi s[J].Art eri oscl er Throm b Vasc Bi ol,2010,3(8): 1513-1518.
[3]Tachi bana H,Ogawa D,M at sushi t a Y,et al.Act i vat i on of l i ver Xrecept ori nhi bi t sost eopont i nandam el i orat esdi abet i c nephropat hy[J].J Am Soc Nephrol,2012,23(11):1835-1846.
[4]St ef f ensen KR,Jakobsson T,Gust af sson JA ,et al.Target i ng l i ver X recept ors i n i nf l am m at i on[J].Expert Opi n Ther Target s,2013,17(8):977-990.
[5]Jayaram an G,Sri ni vas R,Duggan C,et al.P300/cAM P-responsi ve el em ent-bi ndi ng prot ei n i nt eract i ons wi t h et s-1 and et s-2 i n t he t ranscri pt i onal act i vat i on of t he hum an st rom el ysi n prom ot er[J]. J Bi ol Chem,1999,274(24):17342-17352.
[6]Sheppard KA,Rose DW,H aque ZK,et al.Transcri pt i onal act ivat i on by NF-kappaB requi res m ul t i pl e coact i vat ors[J].M ol Cel l Bi ol,1999,19(9):6367-6378.
[7]Li M,Pascual G,Gl ass CK,et al.Peroxi som e prol i f erat or-act ivat ed recept or gam m a-dependent repressi on of t he i nduci bl e ni t ri c oxi de synt hase gene[J].M ol Cel l Bi ol,2000,20(13):4699-4707.
[8]Sem ent chenko VI,W at son DK.Et s t arget genes:past,present and f ut ure[J].Oncogene,2000,19(55):6533-6548.
[9]Xu L,Bai Q,Rodri guez-agudo D,et al.Regul at i on of hepat ocyt e l i pi dm et abol i smandi nf l am m at ory response by 25-hydroxyehol est erol and 25-hydroxychol est erol-3-sul f at e[J].Li pi ds,2010,45(9):821-832.
[10]M aj dal awi eha A,Ro H S.Regul at i on of IkappBal pha f unct i on and NF-kappaB si gnal i ng:AEBP1 i s a novel proi nf l am m at ory m edi at or i n m acrophages[J].M edi at ors Inf l am m,2010,2010(1): 1-27.
[11]Ye J,W ang L,Zhang X,et al.Inhi bi t i on of TNFal pha gene expressi on and bi oact i vi t y by si t e-speci f i c t ranscri pt i on of TNF-al Pha f act or-bi ndi ng ol i gonucl eot i des[J].Am J Physi ol Lung Cel l M ol physi ol,2003,284(2):386-394.
[12]Kunse H C,Ruben SM,Rosen CA.Sel ect i on of opt i m al kappa B/Rel DNA-bi ndi ngm ot i f s:i nt eract i onof bot hsubuni t s of NF-kappa B wi t h DNA i s requi red f or t ranscri pt i onal act i vat i on[J]. M ol Cel l Bi ol,1992,12(10):4412-4421.
[13]W ang Y,Li C,Cheng K,et al.Act i vat i on of l i ver X recept or i m proves vi abi l i t y of adi pose-deri ved m esenchym al st em cel l s t o at t enuat e m yocardi al i schem i a i nj ury t hrough TLR4/NF-kappaB and Keap-1/Nrf-2 si gnal i ng pat hways[J].Ant i oxi d Redox Si gnal,2014,21(18):2543-2557.
[14]Kaur U,Banerj ee P,Bi r A,et al.React i ve oxygen speci es,redox si gnal i ng and neuroi nf l am m at i on i n al zhei m er's di sease:t he NF-kappaB connect i on[J].Curr Top M ed Chem,2015,15(5): 446-457.
[15]Yang SM,Ka SM,W u H L,et al.Throm bom odul i n dom ai n 1 am el i orat es di abet i c nephropat hy i nm i ce vi aant i-NF-kappaB/NLRP3 i nf l am m asom e-m edi at ed i nf l am m at i on,enhancem ent of NRF2 ant i oxi dant act i vi t y and i nhi bi t i on of apopt osi s[J].Di abet ol ogi a,2014,57(2):424-434.
[16]W ang H,Vi nni kov I,Shahzad K,et al.The l ect i n-l i ke dom ai n of t hrom bom odul i n am el i orat es di abet i c gl om erul opat hy vi a com pl em enti nhi bi t i on[J].Throm b H aem ost,2012,108(6):1141-1153.
[17]Chung SW,Kang BY,Ki mSH,et al.Oxi di zed l owdensi t y l i poprot ei n i nhi bi t s i nt erl euki n-12 product i on i n l i popol ysacchari de-act i vat ed m ouse m acrophages vi a di rect i nt eract i ons bet ween peroxi som eprol i f erat or-act i vat edrecept or-gam m aandnucl ear f act or-kappa B[J].J Bi ol Chem,2000,275(42):32681-32687.
[18]Janowski BA,W i l l y PJ,Devi TR,et al.An oxyst erol si gnal l i ng pat hway m edi at ed by t he nucl ear recept or LXR al pha[J].Nat ure,1996,383(6602):728-731.
[19]Fi evet C,St ael s B.Li ver X recept or m odul at ors:ef f ect s on l i pi d m et abol i sm and pot ent i al use i n t he t reat m ent of at heroscl erosi s[J]. Bi ochem Pharm acol,2009,77(8):1316-1327.
(申海菊编辑)
Mechanism and role of LXR agonist upregulating thrombomodulin expression in human glomerular endothelial cells*
Han-lu Ding1,Yi Li1,Yun-lin Feng1,Jin Chen1,Xiang Zhong1,Nan Wang2,Wei Wang1,Ping Zhang1,Li Wang1
(1.Department of Nephrology,Sichuan Provincial People's Hospital,Chengdu,Sichuan 610072,China;2.Department of Nephrology,the Second People's Hospital of Chengdu,Chengdu,Sichuan 610017,China)
ObjectiveToexploretheeffectofliverXreceptor(LXR)agonistT0901317on thrombomodulin(TM)expression in human glomerular endothelial cells(HUGECs)and its mechanism. Methods Western blot was used to detect the expressions of IκBα,p-IκBα,nuclear transcription factor-κB (NF-κβ)p65 and p-NF-κB p65 in HUGECs stimulated by 25 mmol high glucose and 2 μmol T0901317.The interaction between LXR and the transcriptional coactivator p300 in HUGECs was detected using coimmunoprecipitation(Co-IP)assay.The concentrations of IL-1β and TNF-α in the supernatant of HUGECs transfected with or without AdTMshRNA were determined using commercially available ELISA kits.Results T0901317(2 μmol)significantly reduced the phosphorylation of IκBα and NF-κB p65 in the HUGECs stimulated by high glucose(P<0.05).The activity of NF-κB was increased by LXR-α silencing despite thepresence of T0901317.Co-IP revealed up-regulation of LXP and p300 in the HUGECs 24 h after stimulation by 2 μmol T0901317.T0901317 inhibited the secretion of high glucose-induced inflammatory cytokines such as TNF-α and IL-1β in the HUGECs(P<0.05).The levels of TNF-α and IL-1β were increased by TM silencing with AdTMshRNA despite the presence of T0901317,but the differences were not significant(P>0.05).Conclusions LXR agonist increases TM expression and inhibits secretion of inflammatory mediators probably by competitively blocking the interaction between NF-κB and p300 through interaction with p300.
glomerular endothelial cell;liver X receptor;thrombomodulin;nuclear transcription factor-κB
R 692.9;TQ 937
A
1005-8982(2016)05-0001-06
10.3969/j.i s s n.1005-8982.2016.05.001
2015-06-01
*
国家自然科学基金(No:81170680);四川省科技厅课题(No:2013JY0141)
王莉,E-m ai l:scwangl i 62@163.com
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2019年6期)2019-08-13
中成药(2018年10期)2018-10-26
中成药(2018年6期)2018-07-11
天然产物研究与开发(2018年6期)2018-07-09
分析化学(2017年12期)2017-12-25
中成药(2017年8期)2017-11-22
中国卫生标准管理(2015年18期)2016-01-20
安徽医科大学学报(2015年9期)2015-12-16
