
铜离子对混合菌群降解三氯乙烯的影响与机制分析
2016-07-04 07:06:09高艳辉赵天涛邢志林何芝张丽杰彭绪亚
生物工程学报 2016年5期
高艳辉,赵天涛,邢志林,,何芝,张丽杰,彭绪亚
1 重庆理工大学 化学化工学院,重庆 4000542 重庆大学 城市建设与环境工程学院,重庆 400045
铜离子对混合菌群降解三氯乙烯的影响与机制分析
高艳辉1,赵天涛1,邢志林1,2,何芝1,张丽杰1,彭绪亚2
1 重庆理工大学 化学化工学院,重庆 400054
2 重庆大学 城市建设与环境工程学院,重庆 400045
高艳辉, 赵天涛, 邢志林, 等. 铜离子对混合菌群降解三氯乙烯的影响与机制分析. 生物工程学报, 2016, 32(5): 621–634.
Gao YH, Zhao TT, Xing ZL, et al. Effects of copper on biodegradation mechanism of trichloroethylene by mixed microorganisms. Chin J Biotech, 2016, 32(5): 621–634.
摘 要:在三氯乙烯 (TCE) 胁迫条件下,从生活垃圾填埋场覆盖土中富集得到了可高效降解TCE的混合菌群SWA1。考察了铜离子浓度0−15 μmol/L范围内混合菌群对TCE的降解,当铜离子浓度为0.03 μmol/L时,降解速率最大为29.60 nmol/min,降解率达95.75%。此条件下的pmoA和mmoX表达量均达最大值,pmoA的相对表达量 (4.22 E-03) 比mmoX (9.30 E-06) 和LmpH (0) 高3个数量级。在0−0.75 μmol/L和1−15 μmol/L两个铜离子浓度区间,分别出现了TCE降解峰值,高通量测序结果表明,甲基孢囊菌科Methylocystaceae的甲烷氧化菌为优势微生物。随着铜离子浓度提高,混合菌群SWA1生物多样性显著降低。铜离子浓度的变化影响了混合菌群的结构和活性,进而影响了TCE降解机制。当铜离子浓度为0.03 μmol/L时,降解机制包括TCE直接降解和甲烷氧化菌共代谢降解。当铜离子浓度为5 μmol/L时,降解率可达到84.75%。此时,降解机制包括TCE直接降解以及甲烷氧化菌和含苯酚羟化酶菌群的共代谢降解。
关键词:混合菌群,三氯乙烯,关键酶,群落结构,降解机制
Received: August 22, 2015; Accepted: November 30, 2015
Supported by: National Natural Science Foundation of China (Nos. 51378522, 41502328), Fundamental and Advanced Research Projects of Chongqing (No. cstc2015jcyjB0015).
国家自然科学基金 (Nos. 51378522, 41502328),重庆市基础科学与前沿技术研究项目 (No. cstc2015jcyjB0015) 资助。
网络出版时间:2016-01-06 网络出版地址:http://www.cnki.net/kcms/detail/11.1998.Q.20160106.1105.002.html
三氯乙烯 (Trichloroethylene,TCE) 是一种重要的有机溶剂和化工原料,但使用过程中的不当处置会导致TCE泄露和直接排放,严重污染了水体、土壤和大气环境。此外,作为重要的TCE人为源,生活垃圾填埋场在有机物降解过程也产生了大量的氯代烃污染物,并伴随着生物气直接排放至大气中,对生态环境造成了严重威胁[1-3]。生物降解因具有高效性及低成本等特点,被认为是去除TCE等氯代烃类污染物的有效途径[4-7]。而与纯菌降解相比,有污染源富集的混合菌群因具有高耐受和互营养的生物特性,可通过共代谢、直接氧化等多种途径更有效的降解氯代烃污染物[8-12]。
研究表明发孢甲基弯菌Methylosinus trichosporium OB3b可以甲烷为碳源通过甲烷单加氧酶 (Methanemonooxygenase,MMO) 对TCE进行共代谢降解。在甲烷、甲醇等底物存在下,MMO经环氧化过程可将TCE代谢为无机氯[9]。MMO有两种存在形式:一种是与细胞膜结合的具有含铜活性中心的颗粒性甲烷单加氧酶 (Particulate methane monooxygenate,pMMO),它存在于除甲基细胞菌属Methylocella和Methyloferula以外的所有已发现的甲烷氧化菌中[13-14],可在高铜离子浓度 (约4 μmol/L) 条件下表达[15-17]。pMMO由α、β和γ亚基以1∶1∶1的比例组成,以 (αβγ)3三聚体形式存在;编码pMMO的基因簇pmmo由3个功能基因组成,按pmoCAB排列,pmoB、pmoA、pmoC分别编码α亚基、β亚基、γ亚基[16,18-19]。Basu 等[20]从荚膜甲基球菌Methylococcus capsulatus Bath中分离纯化得到pMMO的α (47 kDa)、β (27 kDa)、γ (23 kDa) 亚基和一个63 kDa的蛋白质推测为还原酶 (pMMOR),并明确指出这3个亚基组成的蛋白为羟基化酶 (pMMOH)。另一种是分泌在周质空间中的可溶性甲烷单加氧酶(Soluble methane monooxygenase,sMMO),只有少数Ⅰ型甲烷氧化菌和Ⅱ型甲烷氧化菌如甲基球菌属Methylococcus、甲基单胞菌属Methylomonas、甲基细胞菌属Methylocella、甲基孢囊菌属Methylocystis和甲基弯曲菌属Methylosinus[19-21],在铜离子缺乏 (<0.8 μmol/L)条件下可分泌产生[22]。编码sMMO的基因位于由6个基因组成的操纵子上,形成了大约5.5 kb的基因簇,按mmoXYBZDC排列、分别编码α (MMOHα) (60 kDa)、羟化酶亚基β (MMOHβ) (45 kDa)、调控蛋白B (MMOB)、羟化酶亚基γ (MMOHγ) (20 kDa)、MMOD和还原酶C (MMOR) (39 kDa)[19,23]。mmoX与pmoA分别作为编码sMMO和pMMO的保守基因片段,常用来作为编码sMMO和pMMO的功能标志基因开展研究[19,24]。2013年Susanne等[24]利用mmoX基因的转录水平指示sMMO水平,证实了酸性泥炭生态系统中sMMO是一种活性酶。2013年Sheeja等[25]以pmoA为功能基因通过RT-qPCR技术证实了甲基孢囊菌属Methylocystis strain SB2可以以甲醇作为生长基质实现多种污染物降解。铜离子在MMOs催化降解过程中扮演了重要的角色[22],研究表明铜是合成颗粒性甲烷单加氧酶pMMO的必需金属元素,又是调控sMMO或pMMO表达的开关元件[22,26],因此,铜对甲烷氧化菌酶活性、代谢控制具有重要意义。Mukherjee等[27]研究了多种辅酶因子对TCE降解酶活性的影响,发现与其他辅酶因子(各种金属元素) 相比,铜对TCE降解菌嗜麦芽寡养单胞菌Stenotrophomonas maltophilia PM102所产生的酶活性有显著刺激作用。
前期研究表明,生活垃圾填埋场覆盖层在填埋气长期驯化过程中富集出多种环境功能微生物,包括甲基孢囊菌、微杆菌、金黄杆菌、假单胞菌和芽胞杆菌等[28-31]。有氧条件下,填埋气中高浓度挥发性氯代烃 (1 000−5 000 mg/L)可通过甲烷单加氧酶、苯酚羟化酶 (受苯酚羟化酶基因LmPHs控制)、甲苯加氧酶和一些直接氧化酶等催化降解[32-34]。实验室发现从生活垃圾填埋场覆盖土富集到的混合菌群可以有效降解TCE,且铜离子对混合菌群中非甲烷氧化菌及其群落结构有特殊影响。目前,作为重要的辅酶因子,铜离子对混合菌群中非甲烷氧化菌的TCE降解影响并不清晰,对混合菌群酶活调节和TCE降解调控机制还鲜为报道。基于此,本文全面解析了铜离子浓度对TCE生物降解的影响及其响应机制,包括:1) 以甲烷为碳源,TCE胁迫下从生活垃圾填埋场覆盖土中富集可高效降解TCE的混合菌群,初步分析混合菌群的生长特性;2) 分析TCE降解过程中铜离子对混合菌群生物特性、甲烷氧化及TCE降解的影响;3) 通过实时荧光定量逆转录PCR技术分析铜离子对调控TCE降解关键酶保守基因片段转录表达丰度的影响;4) 利用高通量测序技术探究TCE降解过程不同浓度铜离子条件下微生物群落结构,进而明晰混合菌群TCE降解机制。研究成果以期为深入认识混合菌群的共降解机制和生物强化有机物降解提供理论基础。
1 材料与方法
1.1混合菌群的富集驯化
以重庆长生桥生活垃圾填埋场覆盖土为生物介质,选取4 mm筛下和2 mm筛上覆盖土颗粒100 g,置于500 mL血清瓶后具塞密封,用100 mL甲烷置换瓶中空气,30 ℃密闭驯化2周实现甲烷氧化菌的复壮。称取0.2 g已驯化过的土样放入30 mL NMS[28]培养基中,在30 ℃、170 r/min摇床振荡培养2 h。然后取1 mL培养液作为种子接入含30 mL NMS培养基的135 mL血清瓶中,加盖密封。用已灭菌的医用注射器抽取瓶内一定体积的空气,并注入相同体积的甲烷,使瓶中氧气与甲烷的体积比为1∶1,在30 ℃、170 r/min摇床培养,得到混合菌群SWA1。
1.2色谱检测条件
生物气甲烷和氯代烃 (TCE) 浓度检测均采用川仪SC-6000A气相色谱。生物气检测条件:不锈钢色谱柱TDX 8-12-25 2 m;进样器温度、柱温和TCD检测器温度分别为120 ℃、90 ℃和120 ℃;氮气为载气,载气流速为25 mL/min;进样量0.5 mL。氯代烃检测条件:不锈钢色谱柱GDX-104 2 m;进样器温度、柱温和ECD检测器温度分别为120 ℃、80 ℃和200 ℃;氮气为载气,载气流速为40 mL/min;尾吹气速为10 mL/min;进样量:0.1 mL;基流补偿0.00 nA。
1.3基因组DNA和RNA提取
取一定量菌液离心,弃去上清液,用购自天根生化科技有限公司 (北京) 的细菌基因组DNA提取试剂盒和RNAprep Pure培养细胞/细菌总RNA提取试剂盒提取样品的基因组DNA和总RNA。使用超微量紫外分光光度计 (美国Denovix公司) 检测基因组DNA和RNA浓度。
1.4目的基因转录表达

表1 引物及其核苷酸序列Table 1 Primer features
实验所用引物由上海捷瑞生物工程有限公司合成,引物信息如表1所示。其中F27、R1429用于扩增16S rRNA基因上1 540 bp全序列;引物F-16S rRNA、R-16S rRNA用于扩增16S rRNA内参基因上200 bp的核苷酸序列;引物F-pmoA、R-pmoA用于扩增pmoA目的基因上152 bp的核苷酸序列;引物F-mmoX、R-mmoX用于扩增mmoX目的基因上113 bp的核苷酸序列;引物F-LmpH、R-LmpH用于扩增LmpH目的基因上177 bp的核苷酸序列。RNA提取后,基因组DNA去除反应及逆转录反应参照PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time);qPCR反应液配制参照SYBR®Premix Ex TaqTMII (Tli RNaseH Plus)试剂盒 (大连宝生物公司)。
运用实时荧光定量逆转录PCR (RT-qPCR)确定和量化不同生长条件下混合菌群目的基因转录表达情况。将提取的总RNA进行基因组DNA去除反应,逆转录 (RT-PCR) 合成第一链cDNA,以cDNA为模板通过SYBR染料法分别对16S rRNA内参基因和pmoA、mmoX及LmpH目的基因进行qPCR反应。
基因组DNA去除反应条件:42 ℃,2 min,4 ℃保存。逆转录反应条件:37 ℃,15 min,85 ℃,5 s,4 ℃保存。qPCR反应条件:预变性95 ℃,30 s;变性95 ℃,5 s,退火/延伸62.4 ℃(16S rRNA、pmoA、LmpH),30 s;58 ℃ (mmoX),30 s,39个循环,以0.5 ℃频率从65 ℃增加至95 ℃进行熔解。循环结束后荧光信号被收集,仪器记录下每个样品的最小循环阀值即Ct值,将Ct值带入标准曲线方程来确定目标基因的初始拷贝数,将每个基因温度梯度优化后的PCR产物作为标准品,使用超微量紫外分光光度计检测其浓度来计算每个样品DNA中的目标基因的含量,将标准品以10倍的梯度进行稀释作为qPCR标线模板,来分别定量目的基因的初始拷贝数。每次PCR反应结束后,通过分析熔解曲线来确定扩增的特异性。
标准曲线指标均达到了可以量化目的基因的要求,各指标如下,16S rRNA:R2=1.000,E=97.7%,最低检测限为 (2.310E+4) copies/μL。pmoA: R2=0.999,E=97.8%,最低检测限为 (3.96E+4) copies/μL。mmoX:R2=0.999,E=97.4%,最低检测限为 (4.780E+2) copies/μL。LmpH:R2=0.991,E=93.9%,最低检测限为 (3.44E+1) copies/μL。热循环仪器的原始数据转换成每个样品单位体积菌液的目的基因的拷贝数,然后用细菌中最保守的16S rRNA内参基因来归一化其他目的基因。每个样品均设置3个复管。
1.5高通量测序数据处理
基于目前主流的MiSeq平台对16S rRNA基因高变区序列进行测序,测序区域选择V3+V4区,测序片段为468 bp,测序引物为338F-806R[37-38],使用Trimmomatic、FLASH软件对测序数据进行处理获得干净数据。在Usearch软件平台中使用uparse方法将序列按照彼此相似性为97%分归为许多小组,一个小组为一个OTU,从而得到OTU的代表序列。然后,使用uchime检测PCR扩增中产生的嵌合体序列并从OTU中去除,再用usearch_global方法将优化序列map比对回OTU代表序列,最终得到OTU各样品序列丰度统计表。
2 结果与分析
2.1混合菌群SWA1的生长特性
从垃圾填埋覆盖土中富集得到了混合菌群,命名为SWA1,在铜离子浓度为10 μmol/L和无铜离子的对照条件下,以甲烷为碳源进行连续5次传代培养,评估混合菌群在离位条件下的生长稳定性,结果如图1所示。在连续5次传代过程中,混合菌群SWA1生长特性稳定,经过24 h培养后均达到指数生长期,pH值在7.0左右。因此,从垃圾填埋覆盖土中富集的混合菌群SWA1能以甲烷为碳源,实现连续稳定的离位培养。此外,由图1可知,当铜离子浓度为10 μmol/L时,稳定期OD600维持在0.65左右,高于对照组的0.45,这说明添加铜离子有利于混合菌群的生长。
2.2铜离子浓度对混合菌群SWA1降解TCE的影响
共代谢条件 (甲烷浓度为20%) 下,铜离子对混合菌群SWA1降解TCE的影响如图2所示。铜离子添加量的不同导致了TCE降解率和降解速率的变化。当铜离子浓度为0.03、3、5 μmol/L时,在反应的第3天,TCE降解率就分别达到了89.21%、70.15%和80.85% (图2A)。96 h时,随铜离子浓度的增加出现了两个TCE降解峰值。当c (Cu2+)=0.03 μmol/L时,TCE降解率达到最高95.75%,其TCE降解速率为29.60 nmol/min,是其他实验组的1.2−1.6倍。在铜离子浓度为1−15 μmol/L范围内,当铜离子浓度为5 μmol/L时,TCE降解率达到最高的84.75% (图2B)。由Dong等[39]关于pMMO及pMMO-NADH的研究显示随着铜离子浓度的提高,Methylococcus capsulatus Bath甲烷消耗速率逐渐提高。可以初步推断,铜离子浓度变化影响了混合菌群SWA1中非甲烷氧化菌群的活性,继而影响了整个体系降解TCE的活性。为了更好地理解铜离子对混合菌群SWA1共代谢降解TCE的规律,继续考察了降解过程碳源消耗、关键酶定量分析以及群落结构变化。

图1 混合菌群SWA1生长稳定性Fig. 1 Growth stability of the mixed microbial consortium SWA1.
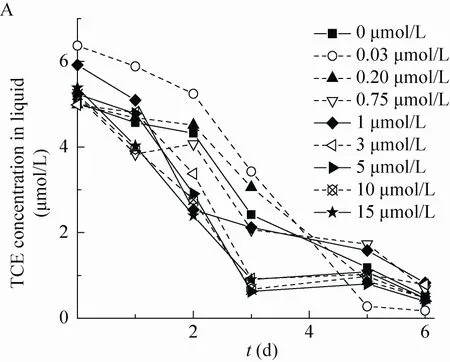
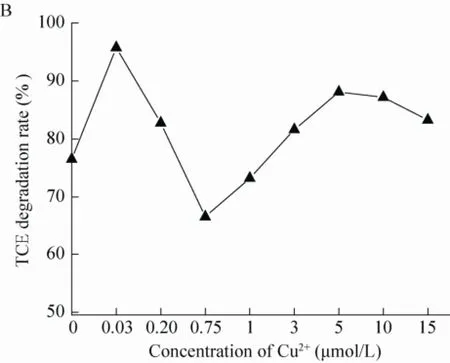
图2 铜离子对混合菌群SWA1生物降解TCE的影响Fig. 2 The effect of copper ion on TCE biodegradation by SWA1.
2.3铜离子对混合菌群SWA1共代谢降解TCE过程甲烷消耗的影响
不同浓度铜离子条件下,TCE降解过程中混合菌群对甲烷的消耗过程如图3所示。空白对照组和铜离子浓度较低 (0.03 μmol/L和0.20 μmol/L)的实验组甲烷消耗速率较低,在反应的第4天甲烷基本完全消耗,比其他实验组延迟了近1 d(图3A)。
考察了各时间段甲烷消耗速率随铜离子浓度变化情况,由图3B可知,随着铜离子浓度的变化,甲烷氧化速率存在着显著差异。反应时间为0−24 h,随铜离子浓度的增加,甲烷氧化速率先增后减,在0.75 μmol/L时,甲烷氧化速率达到最大124.52 mmol/(L·min),当铜离子浓度大于0.75 μmol/L时,铜离子对甲烷氧化的刺激效果逐渐减弱,说明由于短时间内初始高铜离子浓度毒性而产生了不利效果[40]。反应时间为0−48 h和0−72 h,铜离子浓度越大,甲烷氧化速率越快,铜离子的添加显著刺激甲烷的氧化,实验组的甲烷氧化速率是空白对照组的1−2.6倍。这一刺激最可能是由于混合菌群细胞特殊活性的增加引起的,且TCE降解过程中不同铜离子浓度对混合菌群SWA1生长情况的影响也证实了这一点。由图4可知,单位时间内混合菌群细胞浓度随铜离子浓度的增加而增加,同时,稳定期细胞浓度亦与铜离子浓度呈正相关,该现象与Methylococcus capsulatus Bath细胞的浓度及活性随铜离子浓度增加而增加结论相符[39]。此外,72 h平均甲烷氧化速率整体都低于前48 h,但整体趋势一致。这说明随着甲烷逐渐消耗,TCE和甲烷共同竞争关键酶,亦或是TCE氧化产物和酶出现不可逆键合或反应过程还原型辅酶NADH逐渐被消耗。


图3 TCE降解过程中铜离子对甲烷氧化的影响Fig. 3 The effect of copper ion on methane oxidation in the progress of TCE biodegradation by SWA1.
由图3B分析可知,反应时间为0−72 h的区间内,实验组铜离子对甲烷氧化刺激作用总体上均随铜离子浓度增加而增加,平均为对照组的1−1.8倍。以上结果充分说明了铜离子添加促进了甲烷氧化速率的提高,且随反应时间延长铜离子对微生物毒性作用减弱。但与不同铜离子浓度对SWA1生物降解TCE的影响结果(图2) 对比发现,甲烷氧化活性与TCE的降解活性并非正相关,推测和混合菌群中其他微生物菌群的活性存在关联。
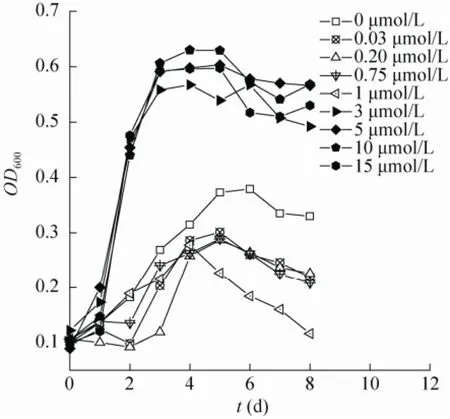
图4 TCE降解过程中铜离子对SWA1生长情况的影响Fig. 4 The growth of SWA1 in the progress of TCE biodegradation by copper ion.
2.4铜离子对混合菌群SWA1关键基因表达丰度的影响
混合菌群对TCE的降解是在sMMO和pMMO等关键酶的催化作用下完成的,TCE降解过程中,不同浓度铜离子条件下混合菌群降解关键酶调控基因转录表达丰度如图5所示。经内参16S rRNA基因归一化后,随铜离子浓度的增加,pmoA和mmoX基因相对表达丰度存在差异,在铜离子浓度为0.03 μmol/L时,pmoA 和mmoX基因表达丰度均出现峰值,而pmoA基因的相对表达量 (4.22E-03±4.98E-05) 比mmoX基因 (9.30E-06±4.89E-7) 高3个数量级,且随铜离子浓度的增加pmoA与mmoX基因相对表达丰度的差异越大,说明该混合菌群富含的甲烷氧化菌主要表达pmoA基因。铜离子在0−0.75 μmol/L浓度区间时mmoX基因的整体表达水平要高于1−15 μmol/L浓度。
Choi等[17]对Methylococcus capsulatus Bath的研究发现,其pmoA基因转录产物的浓度随生长培养基中铜离子浓度 (0−55 μmol/L) 的增加而增加。即铜离子浓度越大,越有利于pmoA基因的表达。而由图5可知,这一规律对于混合菌群SWA1降解TCE的过程并不适用。对照不同铜离子浓度对SWA1生物降解TCE的影响结果 (图2) 也可以发现,TCE的降解并不和MMO丰度呈正相关。以上结果说明随着铜离子浓度变化,混合菌群中非甲烷氧化菌群的活性受到了影响,而这些菌群在TCE降解过程中也起到了关键作用。
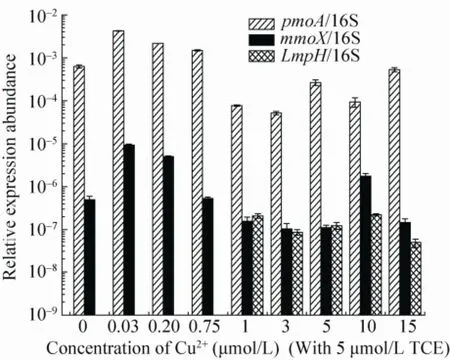
图5 TCE降解过程中不同铜离子浓度对关键基因相对表达丰度的影响Fig. 5 Relative expression abundance of target genes in the progress of TCE biodegradation by copper ion (pmoA/16S: copies of pmoA genes/copies of 16S rRNA genes; mmoX/16S: copies of mmoX genes/copies of 16S rRNA genes; LmpH/16S: copies of LmpH genes/copies of 16S rRNA genes).
已有研究证实苯酚羟化酶等共代谢脱卤酶也可以催化降解TCE[34]。如图5所示,在铜离子浓度为 (0−0.75 μmol/L) 时并未检测到LmpH基因的表达,而当铜离子浓度大于1 μmol/L时该基因开始表达,整体表达水平基本稳定,与mmoX差异不明显,但是与pmoA的表达水平相差2−3个数量级。说明铜离子有利于LmpH基因的表达。而由图2可知,当铜离子浓度为5 μmol/L时,出现了一个TCE降解峰值,这说明在高浓度铜离子区间内,苯酚羟化酶等非甲烷氧化菌的共代谢作用对TCE降解同样起到了关键作用。
2.5铜离子对混合菌群SWA1微生物多样性的影响
TCE降解过程中,在低铜离子浓度和高铜离子浓度范围内分别出现了两个TCE降解峰值,选取该样本进行高通量测序,分析微生物群落结构的变化。铜离子浓度为0.03 μmol/L和5 μmol/L时的混合菌群序列信息和多样性指数如表2所示。两个样品序列数分别为10 799和11 964条,测序的覆盖度基本达到100%,说明对混合菌群中的微生物序列有足够的测序深度,测序结果充分反映了混合菌群中微生物数量和种类的真实情况。
铜离子浓度为0.03 μmol/L和5 μmol/L时的混合菌群微生物群落结构如表3所示,两个样本中优势微生物均为甲基孢囊菌科Methylocystaceae (Ⅱ型甲烷氧化菌) 的甲烷氧化菌。除甲烷氧化菌外,还含有乳球菌属Lactococcus、芽胞杆菌属Bacillus、鞘氨醇单胞菌属Sphingomonas、假单胞菌属Pseudomonas和嗜甲基菌属Methylophilus等多种环境功能微生物。随着铜离子浓度的增加,Methylocystaceae 的OTU数量 (3 899到9 018) 和百分含量(36.10%到75.42%) 显著增大,其他微生物的OTU数量和百分比都显著降低,甚至消失。说明铜离子浓度的增加刺激Ⅱ型甲烷氧化菌的生长,同时对其他非甲烷氧化菌的抑制作用使得高铜离子浓度范围混合菌群的微生物多样性降低,证实了铜离子浓度的变化改变了混合菌群的群落结构。
经调研发现,该混合菌群中非甲烷氧化菌类型的多种微生物都能够进行TCE降解,其OTU数量和生物特性如表3所示。其中乳球菌属Lactococcus、鞘氨醇单胞菌属Sphingomonas能够有效去除TCE,芽胞杆菌属Bacillus能直接以TCE为碳源,节细菌属Arthrobacter和假单胞菌属Pseudomonas含有苯酚羟化酶 (与前面所提到的LmpH的结果相符),能共代谢降解TCE。因此混合菌群SWA1降解TCE是多种途径的协同作用,包括含有MMOs (sMMO和pMMO) 或苯酚羟化酶微生物的共代谢催化氧化,以及芽胞杆菌属、乳球菌属、鞘氨醇单胞菌属等微生物的直接氧化。
由不同铜离子浓度对SWA1生物降解TCE的影响结果 (图2) 可知,铜离子浓度为0.03 μmol/L时,TCE降解率为95.75%,高于另一降解峰值的84.75%。对照不同铜离子浓度对混合菌群SWA1中微生物特性的影响结果(表3),该样本中乳球菌属、鞘氨醇单胞菌属及芽胞杆菌属丰度为13.08%、2.82%和7.94%高于另一峰值样本中的6.29%、1.54%和4.28%。这一结果有力地证明了低浓度铜离子区间,除了共代谢降解作用,直接氧化作用对TCE降解也起到了关键作用。这些微生物之间存在共营养,共代谢等互利共生关系。研究表明Ⅱ型甲烷氧化菌利用丝氨酸途径代谢碳,主要含18-C磷酸脂肪酸,胞内膜分布于细胞壁周围[18],在甲烷氧化菌MMO代谢甲烷及TCE的过程中产生的次级代谢产物如甲醇、甲醛等可能为其他非甲烷氧化菌提供了碳源等,维持了非甲烷氧化菌的存在,共存的非甲烷氧化菌在获得碳源后,产生的一些酶进而可以促进TCE的代谢降解。
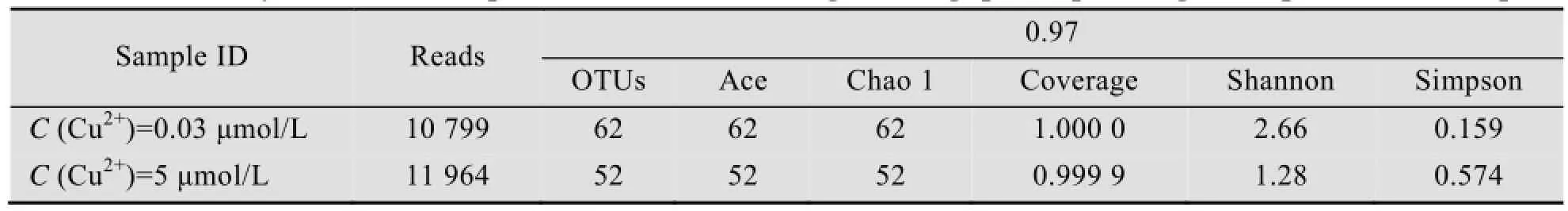
表2 典型样品的高通量测序序列信息和多样性指数Table 2 Diversity indices and sequence information of high throughput sequencing for experimental samples
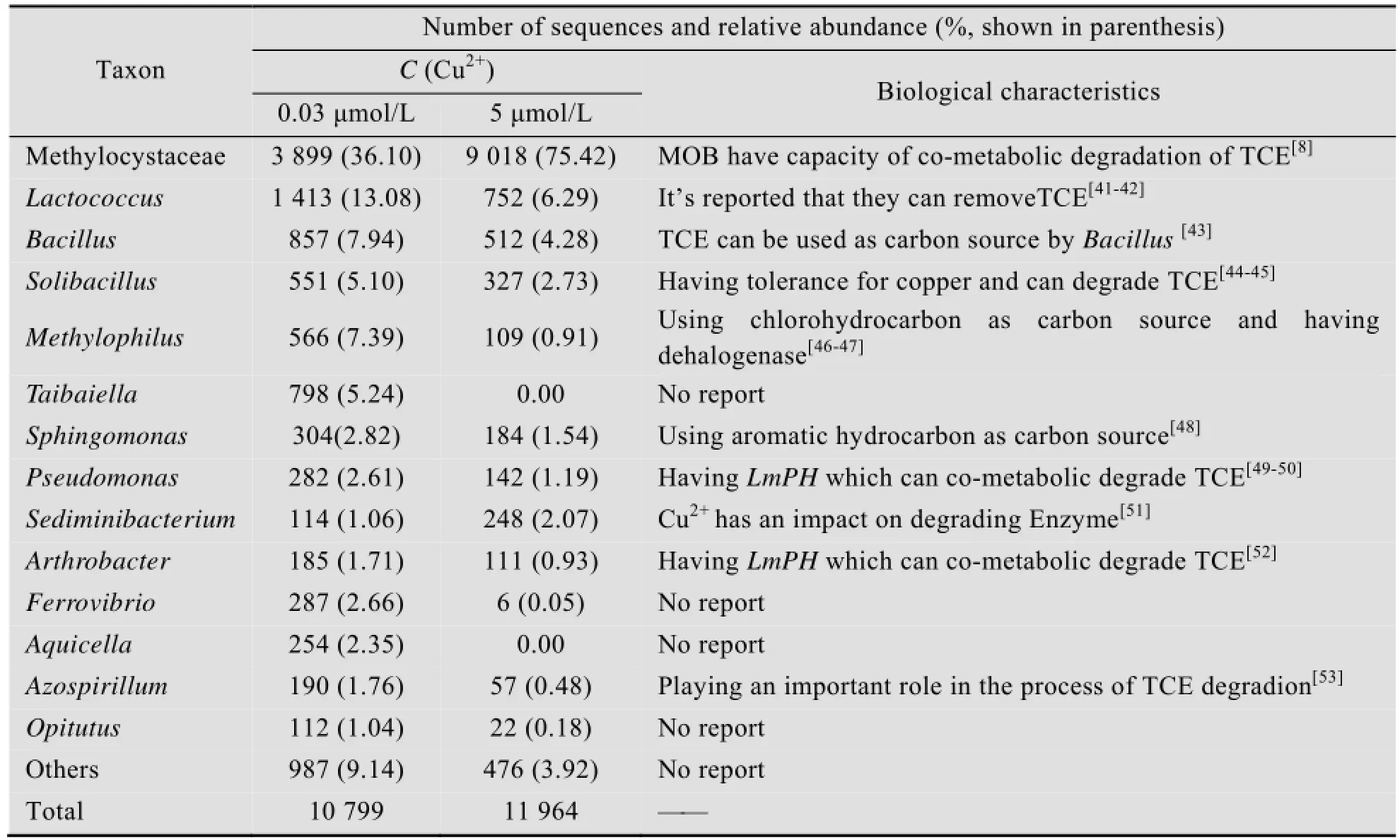
表3 不同铜离子浓度对混合菌群SWA1中微生物特性的影响Table 3 Biological characteristics and number of sequences in genus level of mixed communities under different copper ion concentration
3 结论
以甲烷为碳源,在生活垃圾填埋覆盖土中分离得到了能稳定生长的混合菌群SWA1。该混合菌群对TCE有较强的降解作用,铜离子能够促进其混合菌群生长和TCE降解。pMMO是TCE降解过程中的关建酶,在铜离子浓度为0.03 μmol/L时,pmoA基因和mmoX基因的转录表达丰度出现峰值,添加铜离子有利于LmpH基因的表达。
混合菌群SWA1中优势微生物为甲基孢囊菌科Methylocystaceae的甲烷氧化菌,此外还有乳球菌属Lactococcus和芽胞杆菌属Bacillus等可降解TCE的微生物。铜离子能够改变混合菌群微生物群落结构,由0.03 μmol/L增加到5 μmol/L时,Methylocystaceae比例提高了39.32%,但菌群的生物多样性显著降低。
随着铜离子浓度的变化,混合菌群对TCE的降解存在多种机制。在低浓度区 (0−0.75 μmol/L)主要是MMO的共代谢降解和以TCE作为碳源的直接降解;在高浓度区 (1−15 μmol/L),主要是甲烷氧化菌群和含苯酚羟化酶菌群的共代谢降解。
REFERENCES
[1] Shukla AK, Upadhyay SN, Dubey SK. Current trends in trichloroethylene biodegradation: a review. Crit Rev Biotechnol, 2014, 34(2): 101–114.
[2] Chee GJ. Biodegradation analyses of trichloroethylene (TCE) by bacteria and its use for biosensing of TCE. Talanta, 2011, 85(4): 1778–1782.
[3] Liu JB, Amemiya T, Chang Q, et al. Toluene dioxygenase expression correlates with trichloroethylene degradation capacity in Pseudomonas putida F1 cultures. Biodegradation, 2012, 23(5): 683–691.
[4] Guo Y, Cui KP. Effect of sulfate reduction on biodegradation of trichloroethylene. Chin J Environ Engin, 2014, 8(10): 4159–4162 (in Chinese).郭莹, 崔康平. 硫酸盐还原对三氯乙烯生物降解的影响. 环境工程学报, 2014, 8(10): 4159–4162.
[5] Schmidt KR, Gaza S, Voropaev A, et al. Aerobic biodegradation of trichloroethene without auxiliary substrates. Water Res, 2014, 59: 112–118.
[6] Li Y, Pan T, Liu F, et al. Co-metabolism biodegradation of tetrachloroethylene under different groundwater conditions. Rock Min Anal, 2012, 31(4): 682–688 (in Chinese).李烨, 潘涛, 刘菲, 等. 四氯乙烯在不同地下水环境的生物共代谢降解. 岩矿测试, 2012, 31(4): 682–688.
[7] Paszczynski AJ, Paidisetti R, Johnson AK, et al. Proteomic and targeted qPCR analyses of subsurface microbial communities for presence of methane monooxygenase. Biodegradation, 2011, 22(6): 1045–1059.
[8] Xing ZL, Zhang LJ, Zhao TT. Advances in degradation of chlorinated hydrocarbons by obligate and facultative methanotrophs. Chin J Biotech, 2014, 30(4): 531–544 (in Chinese).邢志林, 张丽杰, 赵天涛. 专一营养与兼性甲烷氧化菌降解氯代烃的研究现状、动力学分析及展望. 生物工程学报, 2014, 30(4): 531–544.
[9] Pant P, Pant S. A review: advances in microbial remediation of trichloroethylene (TCE). J Environ Sci (China), 2010, 22(1): 116–126.
[10] Wang JN, Shi YY, Zheng LY, et al. Isolation and identification of petroleum degradation bacteria and interspecific interactions among four Bacillus strains. Environ Sci, 2015, 36(6): 2245–2251 (in Chinese).王佳楠, 石妍云, 郑力燕, 等. 石油降解菌的分离鉴定及4株芽胞杆菌种间效应. 环境科学, 2015, 36(6): 2245–2251.
[11] Mukherjee P, Roy P. Identification and characterisation of a bacterial isolate capable of growth on trichloroethylene as the sole carbon source. Adv Microbiol, 2012, 2(3): 284–294.
[12] Oldenhuis R, Oedzes JY, Van der Waarde JJ, et al. Kinetics of chlorinated hydrocarbon degradation by Methylosinus trichosporium OB3b and toxicity of trichloroethylene. Appl Environ Microbiol, 1991, 57(1): 7–14.
[13] Dedysh SN, Liesack W, Khmelenina VN, et al. Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bogs, representing a novel subtype of serine-pathway methanotrophs. Int J Syst Evol Microbiol, 2000, 50(3): 955–969.
[14] Vorobev AV, Baani M, Doronina NV, et al.Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int J Syst Evol Microbiol, 2011, 61(10): 2456–2463.
[15] Semrau JD, DiSpirito AA, Yoon S. Methanotrophs and copper. FEMS Microbiol Rev, 2010, 34(4): 496–531.
[16] Zhang YR, Chen HQ, Gao YH, et al. Sequence analysis of 16S rDNA and pmoCAB gene cluster of trichloroethylene-degrading methanotroph. Chin J Biotech, 2014, 30(12): 1912–1923 (in Chinese).张云茹, 陈华清, 高艳辉, 等. 可降解TCE的甲烷氧化菌16S rDNA与pmoCAB基因簇序列分析. 生物工程学报, 2014, 30(12): 1912–1923.
[17] Choi DW, Kunz RC, Boyd ES, et al. The membrane-associated methane monooxygenase (pMMO) and pMMO-NADH: quinone oxidoreductase complex from Methylococcus capsulatus Bath. J Bacteriol, 2003, 185(19): 5755–5764.
[18] Gilbert B, McDonald IR, Finch R, et al. Molecular analysis of the pmo (particulate methane monooxygenase) operons from two type II methanotrophs. Appl Environ Microbiol, 2000, 66(3): 966–975.
[19] McDonald IR, Bodrossy L, Chen Y, et al. Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol, 2008, 74(5): 1305–1315.
[20] Basu P, Katterle B, Andersson KK, et al. The membrane-associated form of methane mono-oxygenase from Methylococcus capsulatus (Bath) is a copper/iron protein. Biochem J, 2003, 369(2): 417–427.
[21] Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol Rev, 1996, 60(2): 439–471.
[22] Green J, Prior SD, Dalton H. Copper ions as inhibitors of protein C of soluble methane monooxygenase of Methylococcus capsulatus (Bath). Eur J Biochem, 1985, 153(1): 137–144.
[23] Han B, Su T, Li X, et al. Research progresses of methanotrophs and methane monooxygenases. Chin J Biotech, 2008, 24(9): 1511–1519 (in Chinese).韩冰, 苏涛, 李信, 等. 甲烷氧化菌及甲烷单加氧酶的研究进展. 生物工程学报, 2008, 24(9): 1511–1519.
[24] Liebner S, Svenning MM. Environmental transcription of mmoX by methane-oxidizing Proteobacteria in a subarctic palsa peatland. Appl Environ Microbiol, 2013, 79(2): 701–706.
[25] Jagadevan S, Semrau JD. Priority pollutant degradation by the facultative methanotroph, Methylocystis strain SB2. Appl Microbiol Biotech, 2013, 97(11): 5089–5096.
[26] Smith SM, Rawat S, Telser J, et al. Crystal structure and characterization of particulate methane monooxygenase from Methylocystis species strain M. Biochemistry, 2011, 50(47): 10231–10240.
[27] Mukherjee P, Roy P. Copper enhanced monooxygenase activity and FT-IR spectroscopic characterisation of biotransformation products in trichloroethylene degrading bacterium: Stenotrophomonas maltophilia PM102. Biomed Res Int, 2013, (6): 643–653.
[28] Zhao T, Zhang LJ, Zhang YR, et al. Characterization of Methylocystis strain JTA1 isolated from aged refuse and its tolerance to chloroform. J Environ Sci, 2013, 25(4): 770–775.
[29] Zhao TT, Xiang JX, Zhang LJ, et al. Screening and biological characteristics of amphitrophic methane-oxidizing bacteria from aged-refuse. Environ Sci, 2012, 33(5): 1670–1675 (in Chinese).赵天涛, 项锦欣, 张丽杰, 等. 矿化垃圾中氧化甲烷兼性营养菌的筛选与生物特性研究. 环境科学, 2012, 33(5): 1670–1675.
[30] Zhao TT, He CM, Zhang LJ, et al. Kinetics of affinity to methane oxidation by Chryseobacterium sp. from aged-refuse. CIESC J, 2011, 62(7): 1915–1921 (in Chinese).赵天涛, 何成明, 张丽杰, 等. 矿化垃圾中金黄杆菌甲烷亲和氧化动力学. 化工学报, 2011, 62(7): 1915–1921.
[31] Song LY, Wang YQ, Tang W, et al. Bacterial community diversity in municipal waste landfill sites. Appl Microbiol Biotechnol, 2015, 99(18): 7745–7756.
[32] Heyer KU, Hupe K, Stegmann R. Methane emissions from MBT landfills. Waste Manag, 2013, 33(9): 1853–1860.
[33] Harborth P, Fuß R, Münnich K, et al. Spatial variability of nitrous oxide and methane emissions from an MBT landfill in operation: strong N2O hotspots at the working face. Waste Manag, 2013, 33(10): 2099–2107.
[34] Galvão TC, Mohn WW, De Lorenzo V. Exploring the microbial biodegradation and biotransformation gene pool. Trends Biotechnol, 2005, 23(10): 497–506.
[35] Luo MF, Wu H, Wang L, et al. Growth characteristics of a methane-utilizing mixed consortia MY9. Chin J Proc Eng, 2009, 9(1): 113–117 (in Chinese).罗明芳, 吴昊, 王磊, 等. 甲烷氧化混合菌群MY9 的生长特性. 过程工程学报, 2009, 9(1): 113–117.
[36] Fierer N, Jackson JA, Vilgalys R, et al. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol, 2005, 71(7): 4117–4120.
[37] Youssef N, Sheik CS, Krumholz LR, et al. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16s rRNA gene-based environmental surveys. Appl Environ Microbiol, 2009, 75(16): 5227–5236.
[38] Hong C, Si Y, Xing Y, et al. Illumina Miseq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas. Environ Sci Pollut Res, 2015, 22(14): 10788–10799.
[39] Prior SD, Dalton H. The effect of copper ions on membrane content and methane monooxygenase activity in methanol-grown cells of Methylococcus capsulatus (Bath). J Gen Microbiol, 1985, 131(1): 155–163.
[40] Ho A, Lüke C, Reim A, et al. Selective stimulation in a natural community of methane oxidizing bacteria: effects of copper on pmoA transcription and activity. Soil Biolog Biochem, 2013, 65: 211–216.
[41] Zhang Y, Hu M, Li PF, et al. Trichloroethylene removal and bacterial variations in the up-flow anaerobic sludge blanket reactor in response to temperature shifts. Appl Microbiol Biotechnol, 2015, 99(14): 6091–6102.
[42] Zhang Y, Wang X, Hu M, et al. Effect of hydraulic retention time (HRT) on the biodegradation of trichloroethylene wastewater and anaerobic bacterial community in the UASB reactor. Appl Microbiol Biotechnol, 2015, 99(4): 1977–1987.
[43] Dey K, Roy P. Degradation of trichloroethylene by Bacillus sp.: isolation strategy, strain characteristics, and cell immobilization. Curr Microbiol, 2009, 59(3): 256–260.
[44] Toribio-Jiménez J, Rodríguez-Barrera MÁ, Lucena MV, et al. Production of biosurfactants by bacteria isolated from a mine tailing zone in southern Mexico and their resistance to heavy metals. African J Bacter Res, 2014, 6(4): 23–31.
[45] Peña-Montenegro TD, Lozano L, Dussán J. Genome sequence and description of the mosquitocidal and heavy metal tolerant strain Lysinibacillus sphaericus CBAM5. Stand Genomic Sci, 2015, 10(1): 2.
[46] Jenkins O, Byrom D, Jones D. Methylophilus: a new genus of methanol-utilizing bacteria. Int J Syst Evol Microbiol, 1987, 37(4): 446–448.
[47] Stourman NV, Rose JH, Vuilleumier S, et al. Catalytic mechanism of dichloromethane dehalogenase from Methylophilus sp. strain DM11. Biochemistry, 2003, 42(37): 11048–11056.
[48] Mera N, Iwasaki K. Use of plate-wash samples tomonitor the fates of culturable bacteria in mercury and trichloroethylene-contaminated soils. Appl Microbiol Biotechnol, 2007, 77(2): 437–445.
[49] Futamata H, Harayama S, Hiraishi A, et al. Functional and structural analyses of trichloroethylene-degrading bacterial communities under different phenol-feeding conditions: laboratory experiments. Appl Microbiol Biotechnol, 2003, 60(5): 594–600.
[50] Movahedyan H, Khorsandi H, Salehi R, et al. Detection of phenol degrading bacteria and Pseudomonas putida in activated sludge by polymerase chain reaction. Iran J Environ Health Sci Engin, 2009, 6(2): 115–120.
[51] Van Der Ha D, Vanwonterghem I, Hoefman S, et al. Selection of associated heterotrophs by methane-oxidizing bacteria at different copper concentrations. Antonie Van Leeuwenhoek, 2013, 103(3): 527–537.
[52] Agarry SE, Durojaiye AO, Solomon BO. Microbial degradation of phenols: a review. Int J Environ Pollut, 2008, 32(1): 12–28.
[53] Shukla AK, Vishwakarma P, Singh RS, et al. Bio-filtration of trichloroethylene using diazotrophic bacterial community. Bioresour Technol, 2010, 101(7): 2126–2133.
(本文责编 陈宏宇)
农业生物技术
Effects of copper on biodegradation mechanism of trichloroethylene by mixed microorganisms
Yanhui Gao1, Tiantao Zhao1, Zhilin Xing1,2, Zhi He1, Lijie Zhang1, and Xuya Peng2
1 College of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing, 400054, China
2 College of Urban Construction and Environmental Engineering, Chongqing University, Chongqing 400045, China
Abstract:We isolated and enriched mixed microorganisms SWA1 from landfill cover soils supplemented with trichloroethylene (TCE). The microbial mixture could degrade TCE effectively under aerobic conditions. Then, we investigated the effect of copper ion (0 to 15 μmol/L) on TCE biodegradation. Results show that the maximum TCE degradation speed was 29.60 nmol/min with 95.75% degradation when copper ion was at 0.03 μmol/L. In addition, genes encoding key enzymes during biodegradation were analyzed by Real-time quantitative reverse transcription PCR (RT-qPCR). The relative expression abundance of pmoA gene (4.22E-03) and mmoX gene (9.30E-06) was the highest when copper ion was at 0.03 μmol/L. Finally, we also used MiSeq pyrosequencing to investigate the diversity of microbial community. Methylocystaceae that can co-metabolic degrade TCE were the dominant microorganisms; other microorganisms with the function of direct oxidation of TCE were also included in SWA1 and the microbial diversity decreased significantly along with increasing of copper ion concentration. Based on the above results, variation of copper ion concentration affected the composition of SWA1 and degradation mechanism of TCE. The degradation mechanism of TCE included co-metabolism degradation of methanotrophs and oxidation metabolism directly at copper ion of 0.03 μmol/L. When copper ion at 5 μmol/L (biodegradation was 84.75%), the degradation mechanism of TCE included direct-degradation and co-metabolism degradation of methanotrophs and microorganisms containing phenol hydroxylase. Therefore, biodegradation of TCE by microorganisms was a complicated process, the degradation mechanism included co-metabolism degradation of methanotrophs and bio-oxidation of non-methanotrophs.
Keywords:mixed microorganisms, trichloroethylene, key enzymes, community structure, degradation mechanism
Corresponding author:Tiantao Zhao. Tel: +86-23-62563617; Fax: +86-23-62563221; E-mail: zhaott@cqut.edu.cn
猜你喜欢
氯碱工业(2022年9期)2022-12-08 08:53:56
实验科学与技术(2017年4期)2017-09-03 10:56:57
热带农业科学(2016年11期)2017-01-21 14:59:21
热带农业科学(2016年11期)2017-01-21 14:57:08
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:33
河北渔业(2015年10期)2015-10-15 11:14:57
河北渔业(2015年7期)2015-07-21 11:06:52
河北渔业(2015年6期)2015-06-24 11:04:37
癌变·畸变·突变(2015年3期)2015-02-27 06:15:10
河北渔业(2015年1期)2015-01-19 16:28:36
