
Structure,characterization,and dynamic performance ofa wet air oxidation catalyst Cu–Fe–La/γ-Al2O3☆
2016-05-30 01:54:34YongliZhangFengPengYanboZhou
YongliZhang ,Feng Peng *,Yanbo Zhou
1 SchoolofChemistry and ChemicalEngineering,South China University ofTechnology,Guangzhou 510640,China
2 SchoolofChemicaland EnvironmentalEngineering,Hanshan NormalUniversity,Chaozhou 521041,China
1.Introduction
Printing and dyeing wastewater contains complex components and has the characteristics ofhigh chroma,high chemicaloxygen demand(COD),and suspended solids[1].The common treatment methods for printing and dyeing wastewater include coagulation[2],membrane separation[3],biologicaldegradation[4],and adsorption[5].However,printing and dyeing wastewater cannot be treated wellby any of the above unit operations alone.Therefore,several unit operations are combined to treat printing and dyeing wastewater,and the cost of wastewater treatment increases.Moreover,because of insuf ficient wastewater treatment in the traditional process,a large amount of sludge is formed,causing secondary pollution.CWAO is a chemicalprocess in which the organic pollutants are decomposed into CO2and H2O by oxidation at high temperatures(125–320 °C)and pressures(0.5–20 MPa)using oxygen or air as the oxidant[6].The CWAOmethod can be used to treat printing and dyeing wastewater over heterogeneous catalysts at high temperatures and pressures with an increase in reaction rate[7],and the released heat during the process can be used to maintain a high temperature of the system.Thus,the CWAO method is ef ficient and inexpensive,and has no secondary pollution.
Among heterogeneous catalysts,γ-Al2O3is widely used because ofa porous and large speci fic surface area[8].Precious metals such as Ptand Pd show high catalytic activity,long service life,and strong adaptability;however,because of high cost,their applications are limited[9,10].Therefore,a catalyst for CWAO comparable to noble metalcatalysts is highly desired.Transition-metal catalysts are inexpensive,and the lattice oxygen in transition-metaloxides can be easily introduced and removed.They are widely used because of a high catalytic activity[11–13].Hua et al.[14]reported that CuO/γ-Al2O3exhibits excellent performance in the degradation of azo dye and veri fied that radicals are crucialfor the degradation ofhydroxyl.Bautista et al.[15]reported that Fe/γ-Al2O3had an excellent performance in the conversion of phenol,and the reaction temperature and initialpH ofphenolsigni ficantly affected the conversion and TOC removal.Rare-earth metals such as La exhibit specialredox properties and have been widely used as catalysts[16,17].Sun etal.[18]reported that La existed on the surface of Ru in the form of La(OH)3,which could not directly enhance the catalytic effect of Ru catalyst,but prompted ZnSO4to form undissolved(Zn(OH)2)3(ZnSO4)(H2O)3.This salt was adsorbed on the surface of Ru catalystand signi ficantly improved the selective catalytic performance of the Ru catalyst.Mazumder et al.[19]reported that in LaAlO3,La2O3entered theγ-Al2O3and combined with Ni to function well and promote the dispersion and stability of the Niinγ-Al2O3,thus exhibiting a better catalytic effect.At present,the mechanism and activation energy of composite “transition-metaland rare-earth”catalysts have been rarely investigated,particularly those of CFLA catalyst have not been reported.
In previous studies,a Cu1Fe1 homogeneous catalyst containing Cu and Fe was developed[20].The Cu–Fe composite showed improved catalytic activity,and the effectof La was higher than Ce[21].In this study,the simulated printing and dyeing wastewater was treated by the CWAO method using a La-modi fied CFLA composite catalyst.The composite catalyst was prepared and characterized,and its dynamic performance was evaluated.Compared with Cu–Fe/γ-Al2O3(CFA)catalyst without La,the activity and stability of the La-modi fied CFLA catalyst were clearly improved.Thus,this study provides a scienti fic basis for the research and development of rare-earth composite catalysts.
2.Experimental
2.1.Materialpreparation
The CWAO experiments were carried out in a reaction kettle(GS type,Weihai,0.5 L).The printing and dyeing wastewater was simulated with a MOsolution,and ithas a theoreticalCOD for MOconcentration of 3000 mg·L-1.Allof the chemicalreagents(copper nitrate(Cu(NO3)2),ferric nitrate(Fe2(NO3)3),lanthanum nitrate(La(NO3)3),MO,pbenzoquinone(BQ),catalase(CAT),and isopropylalcohol(IPA))used in this article were analytically pure.The catalyst supportγ-Al2O3,purchased from Liaoning province ofChina,was clover with a prismatic section diameter of3 mm and a length of5 mm.
2.2.Catalyst preparation
To increase the speci fic surface area and average pore size,the support was pretreated.First,the support was washed with water twice and then washed with distilled water and dried at 105°C for 10 h in an electrothermalblowing drying oven with ventilation.Finally,the sample was calcined at450°C ata heating rate of6°C·min-1for 2 h in a high-temperature electric box furnace.The calcined supportγ-Al2O3was grinded by a simple physical method,and the 1.18–1.40 mm fraction was selected using screens with 12–14 mesh sizes.
A solution of Cu(NO3)2,Fe2(NO3)3,and La(NO3)3(metalcontent,w=6%)in water was prepared,and the ratio of metal elements m(Cu):m(Fe):m(La)was 1:1:2.The solid support and solution were mixed at a mass ratio of 1:2 and dipped for 12 h.The catalyst was drained,dried at105°C for10 h with ventilation,and the above impregnation and drying process was repeated twice.Finally,the sample was calcined at a set temperature and a heating rate of 6 °C·min-1for 3 h in a high-temperature electric box furnace to afford the finalcatalyst.
2.3.Catalystcharacterization
The surface morphology of the catalyst was observed using a scanning electron microscope(SEM,Shimadzu SSX-550,Japan).The size and distribution of the particles were determined using a transmission electron microscope(TEM,Fei Tecnai G2,USA).The phase composition of the catalyst was determined using an X-ray diffractometer(XRD,Bruker D8-Advance,Germany,CuKαradiation,40 kV,and 40 mA).The binding energy of the elements in the catalyst was determined using an X-ray photoelectron spectrometer(XPS,Kratos AXIS Ultra DLD,Shimadzu,Japan)with monochromatic AlKaX-ray radiation source.The metal leaching in the MO solution was determined using an inductively coupled plasma emission spectrometer(ICP,7700s,Agilent,USA).
2.4.CWAO reaction
The MO solution(0.25 L)and 4.0 g·L-1catalyst were added to the reaction kettle.The mixture was stirred at 500 r·min-1for 120 min under an oxygen partialpressure of2.0 MPa and the reaction temperature of200°C.
To con firm the existence of reactive oxygen species such as·O2-,H2O2,and HO·,the scavenger BQ,CAT,and IPAwith a scavenger concentration of 71.4 mg·L-1(5%of MO concentration)was added to MO solution,respectively.They reacted for 20 min at 160°C,and other reaction conditions are the same as the above.The concentration of MO solution was monitored using a 2012PC UV–visible spectrophotometer atan emission peak of465 nm.
In order to study the catalytic wet oxidation dynamics,the COD of MO solution was monitored at the reaction temperatures of140,160,180,200,and 220°C,and the reaction dynamics was evaluated by calculating the COD removalef ficiency and reaction activation energy.
3.Results and Discussion
3.1.La modi fication ofcatalyst
Fig.1 shows the COD removal ef ficiency of MO solution and the metalleaching in MO solution over different catalysts at the reaction temperatures of 200°C.The activity of the catalysts decreased in the following order:CFLA>CFA,and the corresponding total metal(Cu+Fe+La+Al)leaching in the MO solution was 3.8(1.9+1.0+0.4+0.5)mg·L-1and 7.5(4.2+1.8+0+1.5)mg·L-1,respectively.Compared with the CFA catalyst without La,the COD removal ef ficiency of the MO solution increased from 84.5%to 91.4%over the CFLAcatalyst,and the totalmetalleaching forthe catalyst decreased from 7.5 mg·L-1to 3.8 mg·L-1.Thus,the CFLA catalysthad a high activity and stability.
Homogeneous nitrate catalysts,with the total metal(Cu+Fe+La+Al)concentration of 3.8(1.9+1.0+0.4+0.5)mg·L-1,were added to MO solution,so the COD removalef ficiency of MO solution slightly increased from 67.5%(without the catalyst)to 69.2%.As a result,during the CWAO process,the homogeneous catalysis resulted from a small amount of metal leaching by the heterogeneous catalyst,whose contribution to the MO solution degradation could be negligibly low.
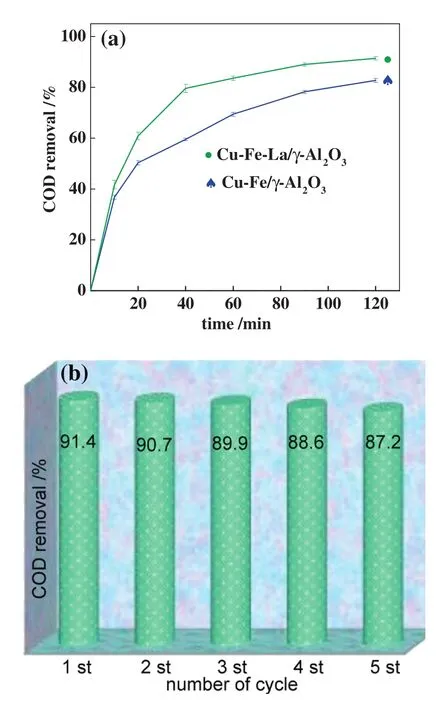
Fig.1.COD removal-time of MO solution over CFA and CFLA catalyst(a)and recyclability of CFLA catalyst(b).
The CFLA catalyst was recycled five times in the CWAO reaction at 200°C and 120 min,and the results are shown in Fig.1.After repeated use,the COD removal ef ficiency of MO solution decreased slightly from 91.4%to 87.2%from the first to the fifth recycle,respectively,which indicated that the catalyst showed higher stability.It exhibited a good cycle performance and reduced the catalytic processing costs,thus improving the utilization rate.
3.2.SEM and TEM analyses
Fig.2(a–d)and(e,f)shows the SEMand TEM images of the CFLA catalyst,respectively.The catalyst calcined at300°C did notcompletely form particles,and the degree of particle aggregation was low(Fig.2(a)),thus decreasing the catalyst stability.The catalyst calcined at 450°C(Fig.2(b))shows a loose structure,uniform particle-size distribution,and uniform distribution ofactive components,which are bene ficialto the adsorption of organic matter;therefore,the catalyst activity increased.Compared with the catalyst calcined at 450°C,the particle size of the catalyst calcined at 600°C clearly increased(Fig.2(c)),and the catalystcalcined at750°C showed a sintered surface due to high calcination temperatures(Fig.2(d)),thus decreasing the catalytic activity.When the calcination temperature was lower than 300°C,the metaloxide could not form particles completely;therefore,the catalysts easily dissolved during the MO solution treatment.With the increase ofcalcination temperature,the particles ofmetaloxides were too dense or sintered,thus affecting the catalytic activity.Therefore,a calcination temperature of450°C was selected.
As shown in Fig.2(e–h),the particle size of the CFLA catalyst Fig.2(e–f)was approximately 10.8 nm,and that of the La-modi fied catalyst Fig.2(g–h)was approximately 7.5 nm with a narrow and even distribution.This was due to the fact that the addition of La impeded the reunion and the growth of the catalyst sample particles.
3.3.XRD analysis
As shown in Fig.3,the XRD peaks for the active components of the catalystappeared at the same positions atdifferentcalcination temperatures,however,the intensity of the XRD peaks changed slightly.The intensity of the XRD peaks increased with increasing calcination temperature,indicating that the sample crystallinity increased.Strong diffraction peaks ofγ-Al2O3appeared at 2θ of 37.6°,39.5°,45.9°,67.0°,and 85.0°,which were attributed to(311),(222),(400),(440),and(444)crystalplanes,respectively.These peaks matched wellwith the reported standard values ofγ-Al2O3(JCPDS 10-0425).The typical diffraction peaks of crystalline Fe2O3(JCPDS 99-060)at 2θ of 24.1°,33.2°,40.9°,54.1°,62.4°,and 64.0°were assigned to(014),(104),(110),(113),(116),and(300)crystal planes,respectively.The dispersed catalyst samples accumulated due to the high calcination temperature of750 °C,and the weaker peak ofγ-Al2O3and other substances strengthened,as detected by the XRD.In addition to the above peak spectrum,the weak diffraction peaks forγ-Al2O3at 2θ of 20.8°,31.9°,and 58.2°were strengthened.The typical diffraction peaks of La2O3(JCPDS 22-0369)at 2θ of 27.2°,31.5°,40.6°,and 72.8°were assigned to(222),(400),(431),and(662)crystalplanes,respectively.The typicaldiffraction peaks of Cu4O3(JCPDS 49-1830)at 2θ of 28.2°and 35.8°were assigned to(112)and(202)crystalplanes,respectively.A part of Cu entered intoγ-Al2O3,forming the spinel CuAl2O4.The diffraction peaks at 2θof 31.2°and 36.8°were assigned to(220)and(311)crystalplanes,respectively.When the calcination temperature was up to 750°C,an increase in the intensity and decrease in the peak width was observed in the XRD patterns during the sintering process,and at the same time the inert substance of CuAl2O4spinelappeared,which severely reduced the catalyst activity.Therefore,the calcination temperature should be lower than 750°C,and the catalyst consisted mainly of Al2O3and Fe2O3,along with a smallamount of La2O3,Cu4O3,and CuAl2O4.

3.4.XPS analysis
The XPS spectra of the CFLA catalyst are shown in Fig.4.Fig.4(a)shows that the main elements in the catalyst were Al,O,Fe,La,Cu,and C.The C element originated from the outside pollution.Fig.4(b)shows that the spectral peak appeared at 530.0,531.2,and 532.4 eV attributed to the peak of the O 1s of lattice oxygen(O2-),adsorbed atomic oxygen(O-),and adsorbed molecular oxygen(O2-),respectively[22].Fig.4(c)shows peaks at 953.7 and 933.9 eV,corresponding to the Cu 2p1/2and 2p3/2of Cu2+.The peaks at 952.5 and 932.9 eV belong to the 2p1/2and 2p3/2of Cu+.Fig.4(d)shows that the peaks at 724.9 and 711.6 eV belong to the 2p1/2and 2p3/2of Fe3+.Peaks which appeared at 724.3 and 710.7 eV belong to the Fe 2p1/2and 2p3/2of Fe2+,respectively.Fig.4(e)shows peaks at 852.1 and 835.8 eV,corresponding to the 3d3/2and 3d5/2spectrallines of La in La3+.The peaks at 855.7 and 839.1 eV can be attributed to the 3d3/2and 3d5/2spectrallines of La,and both of them were strong shake-up satellite peaks.Because La formed composite oxides,La coordinated with the O 2p-valence electrons,resulting in a charge transfer to the empty La 4f orbit.This caused the splitting of the La 3d characteristic peak and generated the shake-up satellite peaks of La 3d3/2and La 3d5/2.Because of the apparent charge transfer between the ligand oxygen and La metal,additionalelectrons existed in the 4f orbit than the initialstate,thus increasing the intensity of the shake-up peaks[23].
The binding energy and concentration ofeach element in the CFLA catalyst are shown in Table 1,and the Cu,Fe,and La elements of the CFLA catalyst mainly existed as Cu2+,Fe3+,and La3+,along with a smallamountof Cu+,Fe2+,and La.
3.5.Mechanism analysis
BQ,CAT,and IPA are the commonly used as scavengers of·O2-,H2O2,and HO·,respectively[24–26].The scavengers are oxidized by ·O2-,H2O2,and HO·,thus decreasing the reaction rate of MO.Fig.5(a)shows the degradation rate of MO by CWAO in the absence ofscavengers BQ,CAT,and IPA,and the mass fraction ofscavenger was an MO of5%,that is 71.4 mg·L-1.Fig.1 shows that the first 20 min ofreaction was accelerated in the CWAO process,thus,20 min reaction time was selected for the degradation of MO.As shown in Fig.5(a),the addition of BQ decreased the degradation rate of MO signi ficantly at 20 min of the reaction.The MO degradation rates of no scavenger,BQ,CAT,and IPA were 59.3%,15.6%,21.2%,and 48.1%,respectively.This shows that·O2-,H2O2,and HO·existed in the MO reaction system for CWAO over the CFLA catalystat the same time.·O2-exhibited the strongestactivity,and H2O2exhibited the centered effect,whereas HO·exhibited the weakest activity.
The possible reaction mechanism is shown in Fig.5(b),where RH is the organic matter and Mis the metal:Cu,Fe,and La.When the CFLA catalyst is used,the possible catalytic oxidation reaction mechanism of MO is shown in Eqs.(1)–(5):

Fig.2.SEMimages of CFLA catalyst calcined at(a)300 °C,(b)450 °C,(c)600 °C,and(d)750 °C;TEMimages and particle size distribution of CFA(e,f)and CFLA(g,h).
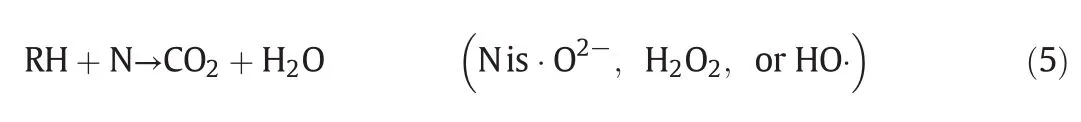
The variable valence states of Cu,Fe,and La endow the CFLA catalyst with excellent electron-transfer properties and catalytic oxidation activity.The low-valence metals capture O2to form·O2-oxidative species[27].Moreover,·O2-combines with H+in the solution,thus affording some stronger oxidative species such as H2O2.The presence ofstrong oxidative species in the CWAO reaction has been an expertconsensus on this issue.In the catalytic oxidation reactions of organic matters,Fu et al.[28]and Wang et al.[29]also con firmed the presence of strong oxidizing species such as·O2-,H2O2,and HO·,which oxidize organic matters into smallermolecules such as CO2,H2O,and other small molecule organic matters.Additionally,according to the literature[30],the N and S elements for the organic matters were primarily decomposed into N2and H2SO3by strong oxidizing species at high temperatures and pressures.

Fig.3.XRD patterns of CFLA catalyst calcined at different temperatures of(a)300°C,(b)450 °C,(c)600 °C,and(d)750 °C.
3.6.Reaction dynamics
According to the CWAO reaction mechanism of MO solution and first-order dynamics[31,32]simulation of the MO degradation,the macro-dynamic Eq.(6)can be expressed as follows:

where rAis the reaction rate;k is the reaction rate constant;and CAis the MO concentration(mg·L-1),and here,denotes the COD(mg·L-1)of MO solution.Eq.(7)can be transformed as follows:


Table 1 Binding energy and concentration ofeach element in the CFLA catalyst
According to the experimental data shown in Fig.6(a),the ln(COD0/COD)under different temperatures and t fits linearly.The fitted straight line slope is the rate constant k of the CWAO reaction for MO solution[33–35].The results are shown in Fig.6(b).According to the fitting results shown in Fig.6(b),ln k was taken as the ordinate,and(RT)-1was taken as the abscissa.Eq.(8)was fitted linearly,and the results are shown in Fig.6(c).


Fig.4.XPS spectra of(a)Survey,(b)O 1s,(c)Cu 2p,(d)Fe 2p,and(e)La 3d over CFLA catalyst.

Fig.5.Effect of scavengers on MO degradation(a)and possible mechanism for MO degradation over CFLA catalyst(b).
As shown in Fig.6,the CWAO reaction for the treatment of the MO solution over the CFLA catalyst can be divided into two stages:the fast reaction period(0–40 min)and the slow reaction period(40–120 min).The large-molecular-weight organics were degraded into low-molecular-weight organic acids in the first stage,which were readily degradable;the low-molecular-weight organic acids were further degraded into carbon dioxide and water in the second stage,which was more dif ficult compared with the former[36].With an extension of the reaction time,the concentration ofstrong oxidizing species in the CWAO reaction reduced;therefore,the apparent activation energy Eaof 13.9 kJ·mol-1in the first section was lower than that of29.5 kJ·mol-1in the second section.The fitting of the two pieces was greater than 0.99,thus,the CWAO reaction of MO solution can be better fitted by a first-order dynamic model.
4.Conclusions
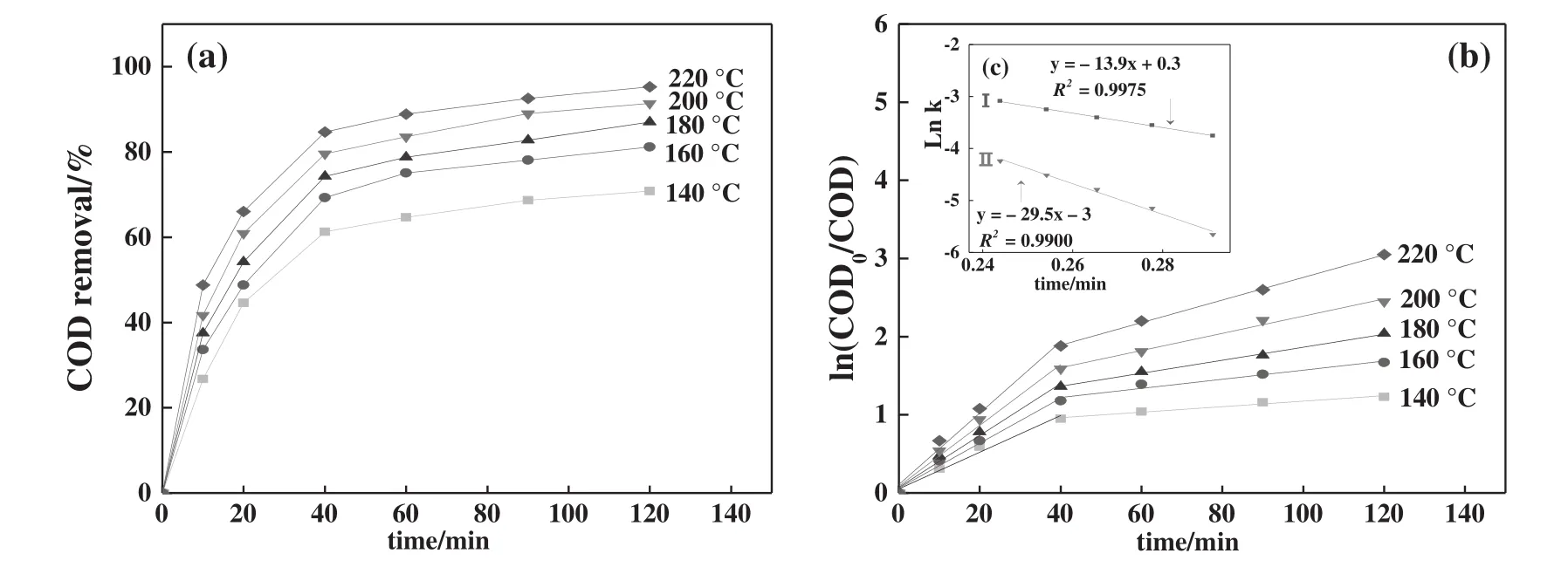
Fig.6.CWAO reaction dynamics of MO solution over CFLA catalyst.
Using the prepared La-modi fied CFLA catalyst,compared with the CFA catalyst without La,the COD removal ef ficiency of MO solution increased from 84.5%to 91.4%by the CWAO method,and the total metal leaching for the catalyst in MO solution decreased from 7.5 mg·L-1to 3.8 mg·L-1.The activity and stability of the Lamodi fied CFLA catalyst improved signi ficantly.The SEM and TEM analyses show that for the catalyst calcined at 450°C,the active components distributed uniformly,and the particle size of the CFLA components was approximately 7.5 nm.The XRD and XPS analyses show that the CFLA components existed as Cu2+,Fe3+,and La3+,along with a smallamount of Cu+,Fe2+,and La.The catalytic reaction of MO solution using the CFLA catalyst involves free-radical reaction mechanism,and the reaction can be fitted by a first-order dynamic model.The apparent activation energy of the fast-phase reaction was 13.9 kJ·mol-1,and the apparent activation energy of the slow-phase reaction was 29.5 kJ·mol-1.
Nomenclature

[1]H.F.Wu,S.H.Wang,Impacts ofoperating parameters on oxidation–reduction potentialand pretreatment ef ficacy in the pretreatment of printing and dyeing wastewater by Fenton process,J.Hazard.Mater.243(2012)86–94.
[2]L.F.Albuquerque,A.A.Salgueiro,J.L.d.S.Melo,O.Chiavone-Filho,Coagulation ofindigo blue presentin dyeing wastewater using a residualbittern,Sep.Purif.Technol.104(2013)246–249.
[3]N.Ben Amar,N.Kechaou,J.Palmeri,A.Deratani,A.Sghaier,Comparison of tertiary treatment by nano filtration and reverse osmosis for water reuse in denim textile industry,J.Hazard.Mater.170(2009)111–117.
[4]J.Fan,H.B.Li,C.D.Shuang,W.T.Li,A.M.Li,Dissolved organic matter removalusing magnetic anion exchange resin treatment on biological ef fluent of textile dyeing wastewater,J.Environ.Sci.26(2014)1567–1574.
[5]T.Santhi,S.Manonmani,T.Smitha,Removal of malachite green from aqueous solution by activated carbon prepared from the epicarp of Ricinus communis by adsorption,J.Hazard.Mater.179(2010)178–186.
[6]M.Martín-Hernández,J.Carrera,M.E.Suárez-Ojeda,M.Besson,C.Descorme,Catalytic wet air oxidation of a high strength p-nitrophenol wastewater over Ru and Pt catalysts:in fluence of the reaction conditions on biodegradability enhancement,Appl.Catal.B Environ.123–124(2012)141–150.
[7]X.J.Hu,L.C.Lei,G.H.Chen,P.L.Yue,On the degradability of printing and dyeing wastewater by wet air oxidation,Water Res.35(2001)2078–2080.
[8]S.de Llobet,J.L.Pinilla,R.Moliner,I.Suelves,Effect of the synthesis conditions of Ni/Al2O3catalysts on the biogas decomposition to produce H2-rich gas and carbon nano fibers,Appl.Catal.B Environ.165(2015)457–465.
[9]T.Nakatsuji,M.Kunishige,J.Li,M.Hashimoto,Y.Matsuzono,Effect of CeO2addition into Pd/Zr–Pr mixed oxide on three-way catalysis and thermal durability,Catal.Commun.35(2013)88–94.
[10]M.Bernardi,M.Le Du,I.Dodouche,C.Descorme,S.Deleris,E.Blanchet,M.Besson,Selective removal of the ammonium-nitrogen in ammonium acetate aqueous solutions by catalytic wetair oxidation over supported Ptcatalysts,Appl.Catal.B Environ.128(2012)64–71.
[11]C.Wang,G.R.Wang,J.F.Wang,A bi-component Cu catalyst for the direct synthesis ofmethylchlorosilane from silicon and methylchloride,Chin.J.Chem.Eng.22(2014)299–304.
[12]P.Massa,F.Ivorra,P.Haure,R.Fenoglio,Catalytic wet peroxide oxidation ofphenol solutions over CuO/CeO2systems,J.Hazard.Mater.190(2011)1068–1073.
[13]F.Fazlollahi,M.Sarkari,H.Gharebaghi,H.Atashi,M.M.Zarei,A.A.Mirzaei,W.C.Hecker,Preparation of Fe–Mn/K/Al2O3Fischer–Tropsch catalyst and its catalytic kinetics for the hydrogenation of carbon monoxide,Chin.J.Chem.Eng.21(2013)507–519.
[14]L.Hua,H.R.Ma,L.Zhang,Degradation process analysis of the azo dyes by catalytic wet air oxidation with catalyst CuO/gamma-Al2O3,Chemosphere 90(2013)143–149.
[15]P.Bautista,A.F.Mohedano,J.A.Casas,J.A.Zazo,J.J.Rodriguez,Highly stable Fe/γ-Al2O3catalyst for catalytic wet peroxide oxidation,J.Chem.Technol.Biotechnol.86(2011)497–504.
[16]A.S.Al-Fatesh,M.A.Naeem,A.H.Fakeeha,A.E.Abasaeed,Role of La2O3as promoter and support in Ni/γ-Al2O3catalysts for dry reforming of methane,Chin.J.Chem.Eng.22(2014)28–37.
[17]M.Ghelamallah,P.Granger,Supported-induced effect on the catalytic properties of Rh and Pt–Rh particles deposited on La2O3and mixed α-Al2O3–La2O3in the dry reforming of methane,Appl.Catal.A Gen.485(2014)172–180.
[18]H.J.Sun,Y.Y.Dong,S.H.Li,H.B.Jiang,Y.Zhang,Z.Y.Liu,S.C.Liu,The role of La in improving the selectivity to cyclohexene ofRu catalystfor hydrogenation ofbenzene,J.Mol.Catal.A Chem.368–369(2013)119–124.
[19]J.Mazumder,H.I.de Lasa,Fluidizable La2O3promoted Ni/γ-Al2O3catalyst for steam gasi fication of biomass:effect ofcatalyst preparation conditions,Appl.Catal.B Environ.168–169(2015)250–265.
[20]Y.L.Zhang,L.Li,X.M.Hu,Preparation ofhomogeneous CWAO catalysts to treat dyeing printing wastewater,J.Northeast Univ.30(2009)1189–1192.
[21]Y.L.Zhang,C.H.Wei,Q.Y.Wang,L.L.Shang,In fluence of preparation conditions on activity and stability of CWAO catalyst Cu–Fe–La/FSC modi fied by La,Res.Environ.Sci.27(2014)210–217.
[22]J.J.Liu,X.M.Jin,W.W.Song,F.Wang,N.Wang,Y.Song,Facile preparation of modi fied carbon black-LaMnO3hybrids and the effect of covalent coupling on the catalytic activity for oxygen reduction reaction,Chin.J.Catal.35(2014)1173–1188.
[23]A.F.Lucrédio,G.T.Filho,E.M.Assaf,Co/Mg/Alhydrotalcite-type precursor,promoted with La and Ce,studied by XPS and applied to methane steam reforming reactions,Appl.Surf.Sci.255(2009)5851–5856.
[24]G.Li,K.H.Wong,X.Zhang,C.Hu,J.C.Yu,R.C.Y.Chan,P.K.Wong,Degradation of Acid Orange 7 using magnetic AgBr under visible light:the roles of oxidizing species,Chemosphere 76(2009)1185–1191.
[25]J.Cao,B.Y.Xu,H.L.Lin,B.D.Luo,S.F.Chen,Chemical etching preparation of BiOI/BiOBr heterostructures with enhanced photocatalytic properties for organic dye removal,Chem.Eng.J.185–186(2012)91–99.
[26]A.C.Bueno,B.B.N.S.Brandão,E.V.Gusevskaya,Aromatization of para-menthenic terpenes by aerobic oxidative dehydrogenation catalyzed by p-benzoquinone,Appl.Catal.A Gen.351(2008)226–230.
[27]W.Liu,Z.H.Ai,L.Z.Zhang,Design of a neutral three-dimensional electro-Fenton system with foam nickelas particle electrodes for wastewater treatment,J.Hazard.Mater.243(2012)257–264.
[28]D.M.Fu,J.P.Chen,X.M.Liang,Wet air oxidation of nitrobenzene enhanced by phenol,Chemosphere 59(2005)905–908.
[29]J.B.Wang,W.T.Fu,X.W.He,S.X.Yang,W.P.Zhu,Catalytic wet air oxidation of phenolwith functionalized carbon materials as catalysts:reaction mechanism and pathway,J.Environ.Sci.26(2014)1741–1749.
[30]L.Oliviero,J.Barbier Jr.,D.Duprez,Wet air oxidation ofnitrogen-containing organic compounds and ammonia in aqueous media,Appl.Catal.B Environ.40(2003)163–184.
[31]T.Dong,J.H.Tong,C.Bian,J.Z.Sun,S.H.Xia,Experimentalstudy and kinetic analysis of oxidant-free thermal-assisted UV digestion utilizing supported nano-TiO2photocatalyst for detection of total phosphorous,Chin.J.Chem.Eng.23(2015)93–99.
[32]D.J.Ma,G.G.Liu,W.Y.Lv,K.Yao,X.D.Zhang,H.H.Xiao,Photodegradation of naproxen in water under simulated solar radiation:mechanism,kinetics,and toxicity variation,Environ.Sci.Pollut.Res.Int.21(2014)7797–7804.
[33]W.Zhao,Y.Guo,Y.Faiz,W.T.Yuan,C.Sun,S.M.Wang,Y.H.Deng,Y.Zhuang,Y.Li,X.M.Wang,H.He,S.G.Yang,Facile in-suit synthesis of Ag/AgVO3one-dimensional hybrid nanoribbons with enhanced performance of plasmonic visible-light photocatalysis,Appl.Catal.B Environ.163(2015)288–297.
[34]A.M.T.Silva,J.Herney-Ramirez,U.Söylemez,L.M.Madeira,A lumped kinetic model based on the Fermi's equation applied to the catalytic wet hydrogen peroxide oxidation of Acid Orange 7,Appl.Catal.B Environ.121–122(2012)10–19.
[35]S.D.S.Asaee,L.Vafajoo,F.Khorasheh,A new approach to estimate parameters of a lumped kinetic model for hydroconversion of heavy residue,Fuel 134(2014)343–353.
[36]J.Guo,M.Al-Dahhan,Kinetics of wet air oxidation of phenol over a novel catalyst,Ind.Eng.Chem.Res.42(2003)5473–5481.
 Chinese Journal of Chemical Engineering2016年9期
Chinese Journal of Chemical Engineering2016年9期
- Chinese Journal of Chemical Engineering的其它文章
- In situ synthesis ofhydrophobic magnesium hydroxide nanoparticles in a novelimpinging stream-rotating packed bed reactor☆
- Enhancing the hydration reactivity ofhemi-hydrate phosphogypsum through a morphology-controlled preparation technology☆
- Synthesis and characterization ofcopolymers ofpoly(m-xylylene adipamide)and poly(ethylene terephthalate)oligomers by melt copolycondensation
- Improvement of CO2 capture performance ofcalcium-based absorbent modi fied with palygorskite☆
- Adsorption behavior ofcarbon dioxide and methane in bituminous coal:A molecular simulation study☆
- Characterization of the adsorption behavior ofaqueous cadmium on nanozero-valent iron based on orthogonalexperiment and surface complexation modeling☆