
C-Ce系化合物相结构稳定性的第一性原理研究
2016-05-28 02:51王兰兰黄福祥高恩强阮海光陈志谦
重庆理工大学学报(自然科学) 2016年4期
王兰兰,黄福祥,高恩强,阮海光,陈志谦
(1.重庆理工大学 材料科学与工程学院,重庆 400054;2.西南大学 材料与能源学部,重庆 400715)
C-Ce系化合物相结构稳定性的第一性原理研究
王兰兰1,黄福祥1,高恩强1,阮海光1,陈志谦2
(1.重庆理工大学 材料科学与工程学院,重庆400054;2.西南大学 材料与能源学部,重庆400715)
摘要:采用第一性原理对C-Ce二元系CCe、C2Ce、C3Ce23种化合物的晶体结构进行优化,并对它们的生成焓、结合能、电子结构、弹性性能及德拜温度进行了计算分析。对生成焓、结合能、德拜温度的计算结果表明:3种化合物的生成焓、结合能均为负值,且C3Ce2的生成焓、结合能值最低依次为-0.858 7 eV/atom,-6.999 0 eV/atom;德拜温度最高,为376 K,具有最强的化合物形成能力。电子结构的分析表明:C3Ce2成键电子中C的2s、2p与Ce的5p、5d杂化作用明显,且具有较强的共价性,结构稳定性最好。体模量(B)、弹性模量(E)、泊松比(ν)、理论硬度(Hv)等计算结果表明:C2Ce 不符合力学稳定性标准;CCe为延性、各向异性;C3Ce2为轻微脆性、各向同性,C3Ce2理论硬度远大于CCe,具有较好的力学稳定性。
关键词:C-Ce系化合物;第一性原理;结构稳定性
近年来,计算和模拟技术的发展为越来越多的理论研究提供了方便。作为一种有效预测材料相稳定性的理论方法,第一性原理从最基础的物理学定律对材料性能进行研究,不依赖任何经验参数,计算迅速、方便、准确,应用范围广,已成为研究热点[1-3]。
稀土元素由于具有独特的4f电子层结构,电负性小,化学活性突出,广泛应用于化学热处理。关于稀土对化学热处理的活化催渗、改善渗层组织性能的作用已有大量实验研究验证[4-5],且稀土渗碳[6]、稀土碳氮共渗[7]、稀土渗硼[8]及稀土复合共渗[9]等很多工艺研究已在生产上取得显著成效。
作为化学热处理中一种常见的稀土添加元素,有关Ce在化学热处理中作用的研究成果已有很多。袁泽喜等[10]在研究20钢气相渗碳时发现:在渗剂中加入CeO2可以显著加快渗碳速率,冶炼时在钢中加入的CeO2对渗碳也起加速作用,但作用小于渗剂中的CeO2。陶小克等[11]在稀土硼钒共渗渗剂中添加CeCl3,发现渗入速度提高了40%以上,且渗层硬度提高到2 400 HV0.2,耐磨性也有很大提高。然而,受稀土材料性质、实验复杂度及测试手段等的限制,这些研究都只侧重工艺方面,在理论方面的研究很少,尤其是在物相分析方面。C-Ce二元系化合物是稀土化学热处理中可能出现的重要化合物,目前对其基本物性的研究报道极少,相图还不完善,缺乏系统的认识,还未有准确的理论研究结果。但对其他领域Ce的化合物却有一定的研究:基于第一性原理赝势平面波方法,湖南大学周惦武等[12]计算分析了Mg-Ce 二元合金系中各相的结构稳定性;赵燕燕[13]利用第一性原理方法,分别对Ag-Ce、Cu-Ce、Ni-Ce二元系化合物进行了系统的研究。本文从理论分析入手,结合实验测试数据,运用第一性原理方法对CCe、C2Ce、C3Ce2三种化合物的电子结构、力学性能、德拜温度等进行了系统的计算研究,重点分析讨论了各相结构的稳定性,以期为稀土化学热处理的物相研究提供若干参考数据。
1计算方法与模型
本文采用平面波赝势密度泛函理论的CASTEP(cambridge sequential total energy package) 总能计算软件包[14]。计算时选取广义梯度近似(general gradient approximation,GGA)的PBE形式来处理交换关联能部分[15],计算精度通过平面波动能截断点(ecut)来控制[16]。价电子选取为C:2s22p2和Ce:4f15s25p65d16s2;Ecut设为410 eV,自洽计算(SCF)收敛精度设为1.0×10-5eV/atom;CCe、C2Ce、C3Ce2布里渊区K点分别设为12×12×12,10×10×10,9×9×9。优化后的晶体结构如图1所示。

图1 优化后的CCe、C2Ce、C3Ce2晶体结构
2计算结果与讨论
2.1晶格参数
为分析方法的合理性,得到更精确可信的计算结果,首先对晶体结构进行优化,优化后的平衡晶格常数列于表1中。与实验值对比发现[17-19]:C3Ce2晶格常数误差最小,为0.4%;CCe 次之,为1.3%;C2Ce晶格常数的误差稍大,为3.3%,主要是因为晶格的热膨胀效应(实验值测量温度一般是室温,此理论模拟的温度为0 K)。总的来说,晶格常数理论模拟结果和实验值非常接近,说明本文采用的相关计算条件和理论模型合理可信。
2.2生成焓和结合能
为研究CCe、C2Ce、C3Ce23种化合物的热力学性能,本文根据式(1)、(2)[20]计算了3种化合物的生成焓ΔH和结合能Ecoh,结果如表2所示。
(1)
(2)

表1 CCe、C2Ce、C3Ce2 的晶体结构、晶格常数及原子分布情况

表2 CCe、C2Ce、C3Ce2 的生成焓和结合能
生成焓可以用来表示物相晶体结构形成的难易程度,生成焓值越小,合金化形成能力越强。结合能用来反映晶体结构的稳定性,结合能的绝对值越大,形成的晶体结构越稳定。
由表2可以看出:三者的生成焓和结合能均为负值,说明三者均有一定程度的热力学稳定性;C3Ce2的生成焓和结合能值均最小,说明C3Ce2合金化形成能力最强,且形成的晶体结构最稳定,C2Ce次之,CCe最差。
2.3电子结构
为研究化合物的电子结构,进一步分析相结构稳定性差异的原因,本文计算了CCe、C2Ce和C3Ce2沿布里渊高对称点方向的总态密度(total density of states,TDOS)和分波态密度(partial density of states,PDOS),结果如图2所示。分析时侧重考虑能量值在0 eV位置(点虚线表示费米面)附近态密度变化情况,横坐标表示能量,纵坐标表示态密度值。
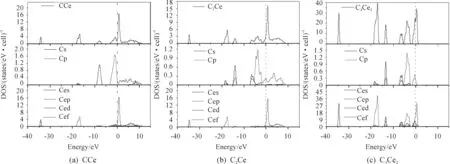
图2 CCe(a)、C2Ce(b)和C3Ce2(c)沿布里渊高对称点方向的总态密度TDOS和分态密度PDOS
由图2可以明显看出:3种化合物在费米能级处的态密度均不为0,均表现出一定的金属性。对比发现3种化合物的态密度图有一定程度的相似,成键电子均主要集中在4个区间:在-40~-30 eV 区域参与成键的电子轨道均为Ce的6 s;在-20~-15 eV区域均主要由Ce的5p轨道成键;费米面以上参与成键的电子轨道均为Ce的4f;在-15~0 eV区域,CCe主要由C的2s、2p与Ce的5d电子杂化成键,C2Ce主要由C的2s、2p作用,C3Ce2主要由C的2s、2p电子与Ce的5p、5d杂化作用成键。杂化意味着更多稳定键的形成,强杂化有益于说明相的稳定性。比较三者的态密度发现,在费米面以下,CCe和C3Ce2的态密度均有不同程度的杂化,且C3Ce2的杂化最强。这说明C3Ce2最稳定,CCe次之,C2Ce几乎没有杂化,稳定性最差。赝能隙可以反映体系成键的共价性的强弱,赝能隙越宽,共价性就越强[21]。对比发现:C3Ce2、CCe、C2Ce态密度图费米能级处成键峰的高度依次降低,表明三者成键电子数依次减少,价电子间相互作用力依次减弱,相稳定性依次降低,且C3Ce2两侧尖峰间距(赝能隙)较大,体现很强的共价性。这些都进一步说明三者结构稳定性顺序为:C3Ce2>CCe>C2Ce。
2.4弹性性能
为研究分析CCe、C2Ce,C3Ce23种化合物的力学性能,分别计算了它们的弹性常数,结果见表3。立方晶系(CCe、C3Ce2)独立的弹性模量有3个,保持稳定性的条件为[22]:
四方晶系(C2Ce)独立的弹性模量有6个,需要满足的条件为[23]:
根据Born-Huang的力学稳定性标准,立方晶系的CCe、C3Ce2符合稳定性条件,四方晶系的C2Ce不符合稳定性条件,因此本文无需再讨论C2Ce的力学性能。
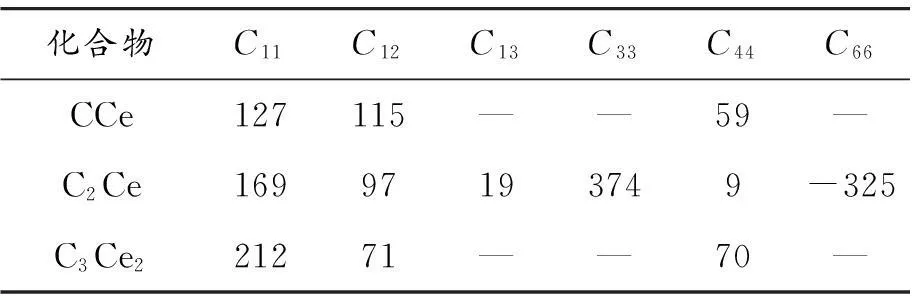
表3 CCe、C2Ce和C3Ce2的弹性常数Cij
体模量B和剪切模量G可根据目前估算多晶体弹性模量最好的Voig-Reuss-Hill近似法[24]获得,计算公式如下:
(3)
(4)
弹性模量(E)和泊松比(v)计算公式如下[25]:
(5)
(6)
作为平均价键强度的标准,体模量B在一定程度上反映了材料抵抗外力的能力,B越大,平均价键强度越大。从表4中可以看出:C3Ce2的B值与CCe的大小相等,这说明二者抵抗外力的能力相当。通常来说,材料的G值、E值与硬度存在一定的正比关系,G值、E值越大,材料的硬度越大。对比发现:C3Ce2的G值、E值明显大于CCe的相应值,说明C3Ce2硬度远远大于CCe。此外,G/B、泊松比ν的大小可以用来评估和预测材料的延性和脆性[26-27]。当G/B<0.57时,表现为延性;G/B>0.57时表现为脆性,且G/B的值越大,脆性越明显。当泊松比ν>1/3时,表现为延性;当ν<1/3时,表现为脆性,ν值愈大,材料的延性愈好。从表4中的计算结果可以看出:CCe的G/B=0.21<0.57,ν=0.4>1/3,表现为延性;C3Ce2的G/B=0.58>0.57,ν=0.2571<1/3,表现为轻微脆性。
通常,理论硬度HV的计算公式为[28]:
(7)
式中:k=G/B;各向异性系数AU可根据Ranganathan和Ostoja-Starzewski提出的各向异性理论[29]由式(8)得出。
(8)
式中GV,BV,GR,BR分别表示由Voigt和Reuss方法得出的剪切模量和体积模量。
作为衡量材料耐磨性的重要指标,硬度是设计耐磨涂层和耐磨材料时不可或缺的力学参数。一般来说,硬度越大,耐磨性越好。仅通过理论计算出来的硬度值虽然和实际硬度值相比存在一定误差,但通过比较,依然可以得出很多有关硬度大小的信息。对比发现:C3Ce2的理论硬度远大于CCe。因此可以猜测:C3Ce2的耐磨性等优于CCe,这和前面的G和E值分析一致。同时,根据二者的各向异性系数可以看出:CCe的各向异性系数与0相差大,为典型的各向异性材料;C3Ce2的各向异性系数为0,为各向同性材料。
2.5德拜温度
作为反映物质结构稳定性、成键原子间化学键结合强度等的一个重要的特征量,德拜温度ΘD还与许多热力学性质紧密相关,因此有必要对其进行研究分析。ΘD可通过弹性常数计算获得,计算方法见式(9)~(12)[30],结果见表5。
(9)
(10)
(11)
(12)
式中:h,k,n,M,ρ分别是普朗克常数(h=6.626×10-34J·s)、玻尔兹曼常数(k=1.381×10-23J/K)、化合物中原子数目、化合物的摩尔质量、密度(ρ=M/V);VD,VT和VL分别是平均声速、纵声速和横声速。

表5 CCe和C3Ce2的平均密度、纵声速、
由表5中德拜温度的计算结果可以看出:CCe的德拜温度为247K;C3Ce2的德拜温度为376K;C3Ce2的德拜温度要明显大于CCe的,由于德拜温度可以预测化合物的共价键强度[31],因此可以得出C3Ce2的共价性强于CCe,这和前面B、G及态密度等的分析结果一致。综上,在3种化合物中,C3Ce2具有最好的力学及热力学稳定性。
3结论
1)CCe、C2Ce、C3Ce2的生成焓和结合能大小顺序一致:CCe 2) 态密度表明3种化合物均表现出一定的金属性,通过对成键电子、杂化、赝能隙、成键峰高度的分析得出三者结构稳定性顺序:C3Ce2>CCe>C2Ce。 3)C2Ce不符合力学稳定性标准。对G/B值、泊松比的研究表明:CCe为延性;C3Ce2为轻微脆性。各向异性系数表明:CCe为各向异性;C3Ce2几乎为各向同性,理论硬度远远大于CCe,具有最好的力学稳定性。 参考文献: [1]周朝彪,冉扬强,吴刚,等.第一性原理研究BiNbO4晶体的电子结构和光学性质[J].原子与分子物理学报,2015,32(4):669-673. [2]李琴,罗洋,叶信宇,等.第一性原理计算在相图计算中的应用研究进展[J].有色金属科学与工程,2015,6(6):37-46. [3]SUNG,DINGJF.First-principlesstudyontheelectronic,magneticandspintransportpropertiesofCU(6bpyNO)Cl2[J].JournalofMagnetism&MagneticMaterials,2015,394:22-26. [4]张金柱,杨宗伦,魏可媛.稀土元素在化学热处理中的催渗和扩散机理研究[J].材料导报,2006,20(F05):223-225. [5]张国良,王鸿春,刘成友.稀土共渗技术在化学热处理中的应用[J].热处理技术与装备,2008,29(3):40-46. [6]李香琪.柴油机活塞销应用稀土渗碳工艺的理论分析[J].科技创新导报,2009(4):44-46. [7]董笑鹏.滴注式甲酰氨低温碳氮共渗在铝挤压模生产上的应用[J].模具工业,2012,38(3):64-67. [8]蔡文俊,卢文壮,王晗,等.TC21钛合金稀土渗硼强化表面组织及性能[J].航空学报,2015,36(5):1713-1721. [9]苏谋,黄福祥,曹登驹,等.Cr12 稀土铬钒共渗覆层组织性能的研究[J].重庆理工大学学报(自然科学),2014,28(2):37-39. [10]袁泽喜,谭平,余宗森,等.氧化铈在钢表面气相渗碳时的催化作用[J].稀土,2001,22(1):27-30. [11]陶小克,董桂霞,彭日升,等.固体法稀土硼钒共渗的研究[J].中国稀土学报,2001,19(2):178-181. [12]周惦武,彭平,胡艳军,等.Mg-Ce化合物相结构稳定性的赝势平面波方法研究[J].稀有金属材料与工程,2006,35(6):871-875. [13]赵燕燕.Ag-Cu-Ni-Ce合金系二元稀土化合物的第一性原理计算研究[D].重庆:重庆理工大学,2014. [14]SEGALLMD,LINDANPJD,PROBERTMJ,etal.First-principlessimulation:ideas,illustrationsandtheCASTEPcode[J].JournalofPhysics-condensedMatter,2002,14(11):2717. [15]PERDEWJP,BURKEK,ERNZERHOFM.Generalizedgradientapproximationmadesimple[J].Physicalreviewletters,1996,77(18):3865-3868. [16]李燕峰,徐慧,张彪,等.Al-Sc金属间化合物的电子结构及稳定性和热力学性质[J].中国有色金属学报,2010,20(5):946-953. [17]STECHERP,NECKELA,BENESOVSKYF,etal.UntersuchungenindenDreistoffenCe-Th(U,Zr,Mo) -C[J].PlanseeberichtefürPulvermetallurgie,1964,12:181-195. [18]JONESDW,MCCOLMIJ,STEADMANR,etal.Neutronpowderdiffractionstudyoftetragonalcrystalstructuresofcerium-neodymiumdicarbides[J].PhaseTransition,1992,38:127-220. [19]ATOJIM,WILLIAMSDE.Neutron-diffractionstudiesofLa2C3,Ce2C3,Pr2C3andTb2C3[J].JournalofChemicalPhysics,1967,47:1188-1189. [20]傅利,赵宇宏,杨晓敏,等.Mg-Al-Si-Ca合金系金属间化合物的电子结构和力学性能的第一性原理计算[J].稀有金属材料与工程,2014,43(11):2733-2738. [21]马振宁,蒋敏,王磊.Mg-Y-Zn合金三元金属间化合物的电子结构及其相稳定性的第一性原理研究[J].物理学报,2015,64(18):187102. [22]BORNM,HUANGK.DynamicalTheoryofCrystalLattices[M].Oxford:ClarendonPress,1954. [23]XUWW,HANJJ,WANGY,etal.First-principlesinvestigationofelectronic,mechanicalandthermodynamicpropertiesofL12orderedCo3(M,W) (M=Al,Ge,Ga)phases[J].ActaMaterialia,2013,61:5437-48. [24]刘洪江,潘勇,管伟明,等.过渡金属硼化物超硬材料的研究进展[J].稀有金属,2013,37(4):633. [25]SCHREIBERE,ANDERSONOL,SOGAN.Elasticconstantsandtheirmeasurements[M].NewYork:McGraw-Hill,1973. [26]HAINESJ,LEGERJM,BOCQUILLONG.Synthesisanddesignofsuperhardmaterials[J].Annualreviewofmaterialsresearch,2001,31(1):1-23. [27]ZHOUP,GONGHR.Phasestability,mechanicalproperty,andelectronicstructureofanMg-Casystem[J].JournaloftheMechanicalBehaviorofBiomedicalMaterials,2012(8):154-164. [28]CHENXQ,NIUHY,LIDZ,etal.Modelinghardnessofpolycrystallinematerialsandbulkmetallicglasses[J].Intermetallics,2011,19(9):1275-1281. [29]RANGANATHANSI,OSTOJA-STARZEWSKIM.Universalelasticanisotropyindex[J].PhysicalReviewLetters,2008,101(5):055504. [30]ANDERSONOL.AsimplifiedmethodforcalculatingtheDebyetemperaturefromelasticconstants[J].JournalofPhysicsandChemistryofSolids,1963,24:909-917. [31]闵婷,高义民,李烨飞,等.第一性原理研究碳化铬的电子结构,硬度和德拜温度[J].稀有金属材料与工程,2012,41(2):271. (责任编辑陈艳) The First-Principle Study on Phase Stability of C-Ce Intermetallic Compounds WANG Lan-lan1, HUANG Fu-xiang1, GAO En-qiang1,RUAN Hai-guang1, CHEN Zhi-qian2 (1.College of Materials Science and Engineering, Chongqing University of Technology,Chongqing 400054, China; 2.Faculty of Materials and Energy,Southwest University, Chongqing 400715, China) Abstract:The crystal structure of CCe, C2Ce, C3Ce23 were geometry optimized by using the first-principle method. On this basis, their enthalpy of formation, cohesive energy, electronic structure, elastic properties and Debye temperature were calculated and analyzed. The calculated results of enthalpy of formation and cohesive energy indicate that all of them have the negative enthalpy of formation and cohesive energy, while C3Ce2have the strongest compound forming ability, of which the enthalpy of formation is -0.858 7 eV/atom, the cohesive energy is -6.999 0 eV/atom, and the Debye temperature is 376 K. The analysis of electronic structure shows that the structural stability of C3Ce2 is best, duing to that the bonding electrons of C3Ce2 have a obvious C(2s),C(2p) and Ce(5p),Ce(5d) hybridization, which means a strong covalence. Bulk modulus(B), Young’s modulus(E), Poisson’s ratio(ν), hardness of theory (Hν) can give the information that C2Ce didn’t meet the mechanical stability conditions, and CCe is ductile and anisotropic, while C3Ce2 presents slight brittleness and isotropic, and theory hardness of C3Ce2 is greater than CCe, which means C3Ce2 has a better mechanical stability. Key words:C-Ce compound; the first-principle method; phase stability 文章编号:1674-8425(2016)04-0046-07 中图分类号:O641 文献标识码:A doi:10.3969/j.issn.1674-8425(z).2016.04.009 作者简介:王兰兰(1990—),女,硕士,主要从事功能材料及材料计算研究;通讯作者 黄福祥,男,博士,教授,主要从事有色金属功能材料、模具表面强化、机械零件失效分析研究。 基金项目:重庆市科技攻关计划项目(cstc2012gg-yyjs50009) 收稿日期:2015-11-16 引用格式:王兰兰,黄福祥,高恩强,等.C-Ce系化合物相结构稳定性的第一性原理研究[J].重庆理工大学学报(自然科学),2016(4):46-52. Citation format:WANG Lan-lan, HUANG Fu-xiang, GAO En-qiang,et al.The First-Principle Study on Phase Stability of C-Ce Intermetallic Compounds[J].Journal of Chongqing University of Technology(Natural Science),2016(4):46-52.
