
MC1R基因编码区的系统发育分析和正选择位点检测
2016-05-03 20:13李丽莎李祥龙周荣艳李兰会张永改
江苏农业科学 2016年3期
李丽莎+李祥龙+周荣艳+李兰会+张永改+马梦云+张秀玲


摘要: 黑素皮质素受体1(melanocortin 1 receptor,MC1R)基因在黑色素细胞的表达上发挥重要作用,并与皮肤色素沉积密切相关。MC1R基因的选择压力分析对识别重要的功能位点具有深远影响。为研究不同动物MC1R基因在进化过程中是否受到正选择作用,基于位点模型对MC1R基因进行研究。结果表明,动物MC1R基因主要经历中性漂变和纯化选择作用,并且发现MC1R基因受到正选择作用的是编码29、191、253、304氨基酸的位点。本研究从分子进化的角度为MC1R基因调控黑色素的形成提供了一个新的思路。
关键词: MC1R;分子进化;亲缘关系;系统发育;正选择
中图分类号: Q756 文献标志码: A 文章编号:1002-1302(2016)03-0044-04
黑素皮质素受体1(melanocortin 1 receptor,MC1R)基因,也称为促黑素细胞激素受体(MSHR)基因,该基因只有1个编码区,由扩展位点(extension locus,E)编码,是控制动物黑色素合成的一个重要基因。哺乳动物的MC1R一般由310个氨基酸组成,有7个跨膜结构域,是G蛋白耦合受体-黑素皮质素受体(melanocortin receptor,MCRs)的家族成员之一[1],该基因在黑色素细胞的表达上发挥重要作用,并与皮肤色素沉积密切相关。MC1R与α-促黑素(α-melanocyte stimulating hormone,α-MSH)和肾上腺皮质激素(ACTH)结合,增强了腺苷酸环化酶的活性,使环腺苷酸水平提高,进而提高酪氨酸激酶(TYR)表达,从而形成黑色素[2]。它们之间的结合使动物毛色或羽色的调控得以正常进行。目前,已知一些MC1R基因变异引起腺苷酸环化酶活化能力不同,并且与小鼠、牛、马、狐狸、家鸡等动物中的皮毛差异有关[3-4]。许多哺乳动物有着同源特征的MC1R基因已被描述,而对于鸟类来讲,MC1R基因色素性状的调控机理与哺乳动物相同[5],因此MC1R基因通过控制真黑色素的数量来控制哺乳动物毛色及鸟类羽色,某些动物疾病的发生也与MC1R基因有关[6]。
正选择(positive selection)也称为达尔文选择(Darwinian selection),是指将具有提高个体适合度的有利突变的等位基因在某个群体中固定下来的选择作用,从而适应不同的生存环境[7]。对正选择的研究能更好地理解生物物种的进化过程。从基因水平上来检测适应性进化,这既有利于揭示生物的进化史,同时加深对该基因的结构和功能变异的理解[8]。
随着对MC1R基因的进一步深入研究,全面检测MCIR基因位点将对动物品种多样性的保护和利用提供依据。对MC1R基因的研究已经数年,传统研究大多集中在MC1R基因的结构、生物学功能和作用机理,往往忽略对其结构、功能和作用机理起决定作用的基因序列的研究。在这些序列中蕴藏着大量的生物信息,对今后MC1R基因的深入研究提供重要依据和参考。本研究对21种动物MC1R基因编码区进行分子进化分析,以期明确该基因在动物演化过程中是否发生了正选择作用,从而更加深入了解MC1R基因。
1 材料与方法
1.1 序列数据
本研究利用已有的MC1R基因序列数据进行研究,所有的数据来源于NCBI网站(http://www.ncbi.nlm.nih.gov/)GenBank数据库,下载20种哺乳动物45条MC1R基因的编码区(963 bp),再加上1条原鸡(AY220303.1 Gallus gallus)序列作为外群,总共21个物种46条MC1R基因序列(表1)。
1.2 方法
1.2.1 序列分析 利用Clustal W 2.1软件对MC1R基因的蛋白序列进行联配,再通过Pal2nal[9]在线工具生成密码子多序列比对,生成序列文件。使用Bioedit 7.0软件[10]对序列进行编辑并分别保存成FASTA格式和PHY格式的文件。用MEGA 6.0软件[11]将比对序列转化成NEXUS格式用于贝叶斯算法来构建系统发育树。利用DAMBE软件[12]进行碱基替代饱和度检验(test of substitution saturation),以确定序列是否适合建树,若建树所用的序列没有达到饱和,则可以建树并进行系统发育分析,若达到饱和则无建树的必要。通过该软件将比对后序列转化成PAML格式用于选择压力分析。
1.2.2 系统发育树的构建 通过PAUP4.0[13]和Modeltest3.7[14]计算出符合该序列的核苷酸最佳替换模型。基于赤池信息量准则(Akailke Information Criterion,AIC)选出的模型为TIM3+I+G,其参数结果用于MrBayes软件[15]的计算,利用贝叶斯算法来构建MC1R基因的系统发育树。贝叶斯算法是基于最大似然法(Maximum Likelihood,ML)和贝叶斯推理法(Bayesian Inference,BI)的系统发生分析程序,其后验概率(posterior probabilities)通过3条热链(heated chain)和1条冷链(cols chain)运行1 000 000代估计,MCMC(Markov Chain Monte Carlo)分析中命令mcmc Samplefreq=100,其含义为每100代保存1棵树,直至average standard deviation of split frequencies值小于0.01(该值代表2个独立分析当前的相似程度)可认为达到平衡。丢弃老化样本(sump burnin=2 500)后由剩余样本来构建合意树(Majority consensus tree,Bayesian tree)。获得的系统发育树可通过Treeview软件[16]来观看。
1.2.3 正选择位点的检测 Codeml程序在估算蛋白质编码序列同义替换和非同义替换速率以及检测序列是否经受正选方面被广泛使用。本研究利用Codeml程序来进行位点模型(site models)分析,以检测基因在进化过程中所经受的选择压力的变化[13]。位点模型都是基于选择压力参数ω(编码蛋白质的核苷酸的非同义突变dN与同义突变dS的比值)来衡量分子进化受到选择压力的方向和量度。若ω>1,即非同义突变被固定的速率远远大于同义突变,表明该位点受到正选择压力作用,位于正选择压力作用下的变异有利于新变异体的生存,同时该密码子被称为正选择位点[17-19],而ω=1和 ω<1 分别表示基因在进化过程中经历了中性选择和负选择。其中,位点模型[20-21]假设不同位点的进化速率不同,但在系统发育树的不同分枝上没有差别。参考以下6种模型:(1)模型M0(单一速率模型)假设不同位点进化速率相同;(2)模型M1a(中性选择模型)假设存在一部分保守位点,此时ω<1且其在序列中所占比例为p0,另一部分为中性选择位点,此时ω=1且其在序列中所占比例为p1=1-p0;(3)模型M2a(正选择模型)是在M1a模型的基础上加入正选择位点的比例即p2=1-p1-p0;(4)模型M3(离散模型)假设p0、p1和p2分别表示负选择、中性选择和正选择,其对应的选择压力参数分别是ω0、ω1和ω2;(5)模型M7(Beta分布模型)假设0<ω<1;(6)模型M8(Beta-ω分布模型)假设一部分位点的ω>1。利用似然比检验(Likelihood ratio test,LRT)来比较零假设和备择假设模型:M0与M3、M1a与M2a 和M7 与M8的最大似然值对数差的2倍(2ΔlnL),再由改良后的χ2分布进行似然比检验,由P值来判断备择假设是否成立。
2 结果与分析
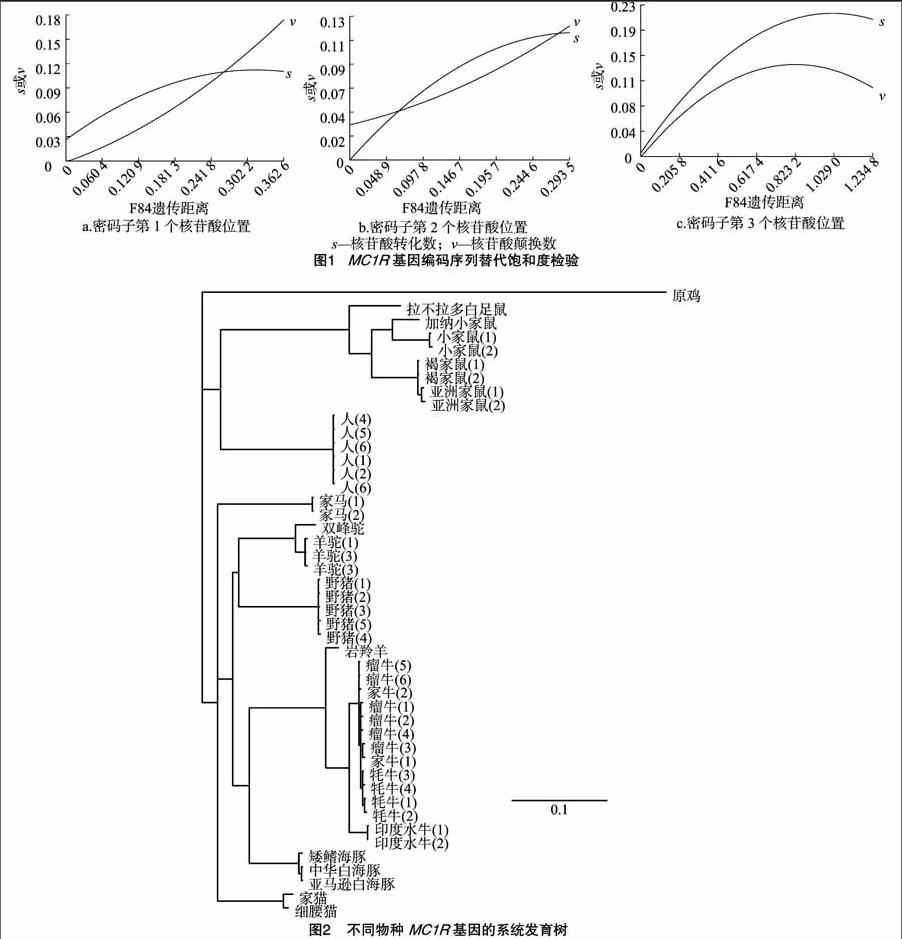
2.1 不同物种MC1R基因的碱基替代饱和度检验
因为进化中转换发生的频率要高于颠换,更容易经受替换和饱和效应的影响,并且饱和效应会直接影响到系统发育分析结果的可信度[22],所以在系统发育分析中检测碱基替代的饱和度是至关重要的。因为编码序列中的三联体密码子第2个编码位置通常是保守的,而第3个密码子位置具有兼并性且对核苷酸同义替换有很大的贡献。普遍认为这是中性理论的微观证据。如果第3个密码子位置处替代达到饱和度,那么序列将会失去进化信号,则以此编码序列来研究基于进化机制的结论可信度不高[23]。本研究使用DAMBE软件在F84模型下检测碱基替代的饱和性,分别利用密码子3个位置处核苷酸转化数(s)和颠换数(v)对遗传距离作图,从图1可以看出,结果均接近线性回归。表明碱基替换未达到饱和,因此可以进行系统发育分析。
2.2 不同物种MC1R基因的系统发育分析
本研究在GenBank数据库进行搜索和筛选,共获得21种动物46条MC1R基因的编码区序列:小家鼠、牦牛、人、野猪、家马和家马等等。利用贝叶斯方法构建系统发育树,利用原鸡的MC1R基因编码区序列作为外类群,建树结果见图2。
由MC1R基因构建的系统发育树主要分成2大分支。一支为牦牛、瘤牛、家牛、印度水牛、野猪、家马、岩羚羊、羊驼、双峰驼、中华白海豚、亚马逊白海豚、矮鳍海豚、家猫、细腰猫14种物种,其中瘤牛、牦牛、家牛、印度水牛、岩羚羊、中华白海豚、亚马逊白海豚和矮鳍海豚8个物种之间亲缘关系较近,而家马、羊驼、双峰驼、野猪、家猫和细腰猫6个物种间亲缘关系较近;另一支为小家鼠、加纳小家鼠、亚洲家鼠、褐家鼠、拉布
拉多白足鼠、人6种物种,其中小家鼠、加纳小家鼠、亚洲家鼠、褐家鼠和拉布拉多白足鼠5个物种之间亲缘关系较近。原鸡与哺乳动物之间区别明显。由此可见,动物MC1R基因构建的系统发育树能很好的反应不同物种之间的亲缘关系。
2.3 正选择位点分析
通过MrBayes软件构建动物MC1R基因的系统发育树后,利用PAML 4软件分析MC1R基因在进化过程中所经受的选择压力(表2)。第1对模型M0和M3似然比检验结果显示2ΔlnL=169.42,df=4,P<0.01。因此模型M3显著优于模型M0,说明位点间承受的选择压力具有异质性。模型M3的3类密码子位点所对应的ω值分别为0.035 30,0.279 60和31.853 85,由于ω2对应的p2为0,因此不存在正选择位点。第2对模型M1a和M2a似然比检验结果显示 2ΔlnL=0,df=2,P=1.00。因此备择假设模型M2a不成立,但并不推荐该模型为参考标准。第3对模型M7和M8似然比检验结果显示2ΔlnL=6.18,df=2,P=0.045。因此备择假设模型M8成立,其ω=1.501 4>1,表明MC1R基因受到选择压力,存在正选择位点(29W,191L,253L,304L)。
本研究用于检测正选择位点的3对模型发现MC1R基因编码的氨基酸位点在进化过程中所经受中性选择和纯化选择作用比例较高,但有4个氨基酸进化受到正选择作用的驱动。
3 讨论与结论
在进化过程中,基因的复制过程所产生的遗传变异会造成功能分歧,随后纯化选择将功能固定下来。少数位点甚至单个位点的核苷酸替代都能改变其编码的蛋白质功能。而整个基因大多数位点都处于纯化选择或中性选择的进化状态,以至于少数位点的正选择信息会被大多数位点所稀释,从而导致正选择信息被整体湮没[24]。现代分子适应性进化分析多以dN/dS的比值来衡量选择压力。若ω>1,且两两模型之间差异显著,则表明编码序列在对应的位点经受正选择作用。与传统方法相比,这种基于最大似然法所估计的正选择检测方法,可把长期由纯化选择和中性选择作用掩盖下的正选择位点识别出来,反应出更高的灵敏度[25]。
本研究采用位点模型来检测选择压力,结果表明,在21种动物中只检测到了4个正选择位点,可见MC1R基因编码的蛋白从整体水平上主要受中性漂变和纯化选择的作用。推测可能是由以下2个原因:(1)MC1R基因所编码的蛋白功能重要,氨基酸序列较为保守。(2)MC1R基因序列进化历史久远,且其较短的序列只能提供较少的可检测位点,这样也难以检测出适应性进化信号[26]。
适应性进化从整体水平上了解了动物MC1R基因在进化过程中所经受的选择作用,同时也分析了不同物种之间亲缘关系的远近。对于亲缘关系较近的物种,选择压力小;而亲缘关系较远的物种选择压力较大且基因序列较保守。MC1R基因作为控制动物毛色形成的主效基因,经证实其变异可作为许多哺乳动物黑、红毛色性状的变化的主要原因[27],该基因功能改变会表现出褐黑色素的红色或黄色性状,反之会表现出真黑色素的黑色或棕色性状[28]。
本研究针对MC1R基因编码区进行分子进化分析,发现MC1R基因编码的蛋白从整体水平上主要受中性漂变和纯化选择的作用,对于正选择作用的分析,共检测到4个正选择位点,这些位点对探讨MC1R基因编码蛋白功能的改变提供了重要依据,从分子进化的角度为MC1R基因调控黑色素的形成提供了一个新的思路。
参考文献:
[1]甘海云,李建斌,王红梅,等. 黑素皮质激素受体1基因与牛的毛色[J]. 家畜生态学报,2007,28(3):99-102.
[2]杨永升,李 宁,邓学梅,等. 黑素皮质素受体1——哺乳动物黑色素形成中的关键基因[J]. 遗传,2004,26(4):544-550.
[3]Sazanov A,Masabanda J,Ewald D,et al. Evolutionarily conserved telomeric location of BBC1 and MC1R on a microchromosome questions the identity of MC1R and a pigmentation locus on chromosome 1 in chicken[J]. Res Dec,1998,8:651-654.
[4]Hunt G,Rot H J,van Oost B A. Identification of a premature stop codon in the melanocyte-stimulating hormone receptor gene(MC1R) in Labrador and Golden ret rievers with yellow coat color[J]. Animal Genetics,2000,31(3):194-199.
[5]Boissy R E,Nordlund J J,Rheins L A. Molecular basis of congenital hypopigmentary disorders in humans:a review[J]. Pigment Cell Research,1997,10:21-24.
[6]Leal-Klevezas D S,Martínez-Vázquez I O,López-Merino A,et al. Single-step PCR for detection of Brucella spp. from blood and milk of infected animals[J]. Journal of Clinical Microbiology,1995,33(12):3087-3090.
[7]Shimizu K K,Purugganan M D. Evolutionary and ecological genomics of arabidopsis[J]. Plant Physiology,2005,138(2):578-584.
[8]Nei M,Kumar S. Molecular evolution and phylogenetics[M]. New York:Oxford University Press Inc,2000.
[9]Suyama M,Torrents D,Bork P. PAL2NAL:robust conversion of protein sequence alignments into the corresponding codon alignments[J]. Nucleic Acids Research,2006,34:W609-W612.
[10]Hall T A. Bioedit:a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT[J]. Nucleic Acids Symp Ser,1999,41:95-98.
[11]Tamura K,Dudley J,Nei M,et al. MEGA4:molecular evolutionary genetics analysis(MEGA)software version 4.0[J]. Molecular Biology and Evolution, 2007,24:1596-1599.
[12]Xia X,Xie Z. DAMBE:data analysis in molecular biology and evolution[J]. Journal of Heredity,2001,92:371-373.
[13]Yang Z H. PAML 4:phylogenetic analysis by maximum likelihood[J]. Molecular Biology and Evolution,2007,24(8):1586-1591.
[14]Posada D,Crandall K A. MODELTEST:testing the model of DNA substitution[J]. Bioinformatics,1998,14(9):817-818.
[15]Ronquist F,Huelsenbeck J P. MrBayes 3:bayesian phylogenetic inference under mixed models[J]. Bioinformatics,2003,19(12):1572-1574.
[16]Page M R D. TreeView:an application to display phylogenetic trees on personal computers[J]. Computer Applications in the Biosciences,1996,12(4):357-358.
[17]Choisy M,Woelk C H,Gue G J,et al. Comparative study of adaptive molecular evolution in different human immunodeficiency virus groups and subtype[J]. Journal of Virology,2004,78(4):1962-1970.
[18]Zhang C Y,Ding N,Chen K P,et al. Complex positive selection pressures drive the evolution of HIV-1 with different co-receptor tropisms[J]. Science China:Life Sciences,2010,53(10):1204-1214.
[19]许慧琳,张文彤,赵耐青,等. H2N2亚型人甲型流感病毒HA1序列进化正选择位点研究[J]. 中华流行病学杂志,2007,28(4):385-389.
[20]Nielsen R,Yang Z. Likelihood models for detecting positively selected amino acid sites and application to the HIV-1 envelope gene[J]. Genetics,1998,148(3):929-936.
[21]Yang Z. Maximum likelihood estimation on large phylogenies and analysis of adaptive evolution in human influenza virus A[J]. Journal of Molecular Evolution,2000,51(5):423-432.
[22]吕雪梅,王应祥,张亚平. 蜂猴线粒体细胞色素b基因变异特点及系统发育分析[J]. 动物学研究,2001,22(2):93-98.
[23]梁国明,刘桂琼,姜勋平. 反刍动物PRNP基因进化机制研究[C]//中国畜牧兽医学会养羊学分会全国养羊生产与学术研讨会议论文集,2010.
[24]姬广超,王明辉,高会江,等. 哺乳动物MT基因的进化选择与功能分歧[J]. 东北农业大学学报,2010(9):82-88.
[25]Yang Z H. Inference of selection from multiple species alignments[J]. Current Opinion in Genetics & Development,2002,12(6):688-694.
[26]Yang Z. Computational molecular evolution[M]. New York:Oxford University Press,2006.
[27]Schmutz S M,Moker J S,Berryere T G,et al. An SNP is used to map MC1R to dog chromosome 5[J]. Animal Genetics,2001,32(1):43-44.
[28]Newton J M,Wilkie A L,He L,et al. Melanocortin 1 receptor variation in the domestic dog[J]. Mammalian Genome,2000,11(1):24-30.
