
Prostatic sarcoma of the Ewing family in a 33-year-old male-A case report and review of the literature
2016-04-27 06:33LuksEschDimitriBrskiReinholdBugThomsOtto
Asian Journal of Urology 2016年2期
Luks Esch,Dimitri Brski,*,Reinhold Bug,Thoms Otto
aDepartment of Urology,Sta¨dtische Kliniken Neuss,Lukaskrankenhaus GmbH,Neuss,Germany
bDepartment of Pathology,Sta¨dtische Kliniken Neuss,Lukaskrankenhaus GmbH,Neuss,Germany
Prostatic sarcoma of the Ewing family in a 33-year-old male-A case report and review of the literature
Lukas Escha,Dimitri Barskia,*,Reinhold Bugb,Thomas Ottoa
aDepartment of Urology,Sta¨dtische Kliniken Neuss,Lukaskrankenhaus GmbH,Neuss,Germany
bDepartment of Pathology,Sta¨dtische Kliniken Neuss,Lukaskrankenhaus GmbH,Neuss,Germany
Ewing sarcoma;
Pelvic neoplasms;
Adult;
Surgery;
Chemotherapy;
Prostate
Ewing sarcoma is the second most common primary bone tumor seen in children and adolescents,typically presenting between 10 and 20 years of age.Extraosseous sarcomas of the Ewing family in adults are rare.We report a manifestation of this tumor entity in the periprostatic tissue of a 33-year-old male and discuss our treatment approach.Transrectal biopsy is a feasible and simple diagnostic tool for unclear pelvic masses.Multi-modal therapy and central registries are needed to gain knowledge of rare pelvic tumors like Ewing sarcoma.
ⓒ2016 Editorial Office of Asian Journal of Urology.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/4.0/).
1.Introduction
Ewing sarcoma is the second most common primary bone tumor seen in children and adolescents.However, extraosseous sarcomas of the Ewing family in adults are very rare.We report a manifestation of this uncommon tumor entity in the periprostatic tissue of a 33-year-old male.
2.Case presentation
A 33-year-old patient who had recently migrated from Afghanistan presented to the emergency department with symptoms of pelvic pain and urinary tract infection (dysuria,urgency,mild pyuria on dipstick)for several days. Patient’s past medical history was unremarkable and without any recollection of exposure to toxic substances or genetic predisposition.Upon suspicion of a pelvic mass on transrectal ultrasound during initial assessment,magnetic resonance imaging(MRI)of the pelvis was performed.It revealed a 6.0 cm×4.5 cm×4.6 cm mostly solid pelvic mass showing signs of central necrosis located between bladder and rectum with suspected infiltration of the prostate and left internal obturator muscle(Fig.1).

Figure 1The 6.0 cm×4.5 cm×4.6 cm mostly solid pelvic mass on T1-weighted,contrast-enhanced(A)as well as T2-weighted MRI-study(B).It shows signs of central necrosis and displacement of bladder and rectum with suspected infiltration of the prostate and left internal obturator muscle;partly contrast-filled bladder with placed foley catheter.
In order to complete tumor-staging,additional cranial MRI,chest/abdominal computed tomography(CT),bonescan as well as colonoscopy were performed.Besides unspeci fic enlargement of a cervical lymph node on the left hand side,no signs of metastatic disease were detected.
For further histological evaluation of the mass, ultrasound-guided transrectal biopsies were obtained. Upon pathological examination the tumor was preliminarily described as a“small,round and blue-cell-like tumor”of the periprostatic soft-tissue in filtrating the capsule of the prostate without further classi fication(Fig.2).
Besides the Ewing’s sarcoma family of tumors,the initial differential diagnoses included desmoplastic round cell tumor and neuroendocrine carcinoma.Other possible differential diagnoses,such as neuroblastoma,rhabdomyosarcoma as well as non-Hodgkin’s lymphoma[1-3],were excluded by immunohistochemistry.With a symptomatic aggressive tumor and in order to get the complete histology,an initial surgical resection was chosen.Radical tumor resection with prostatovesiculectomy,extended regional lymph node dissection as well as resection of the pelvic floor were performed using an open retropubic approach. The bladder neck was then closed and a cystostomycatheter was inserted for urine-diversion(Fig.3).
Course of postoperative recovery was unremarkable.The intraoperative setting suggested at least an R1-situation, even though surgical margins were later reported as tumorfree(R0)by pathology.After further molecular pro filing by a reference pathologist(Prof.Leuschner,Kiel,Germany) the tumor was shown to hold genetic alterations of the EWSR1-gene consistent with the tumor of the Ewing sarcoma family,even though cytokeratin positivity as expressed by this tumor is a rather uncommon histopathological feature only found in a minority of cases in this entity[4,5].Due to the aggressive nature of this disease and given the likely presence of residual disease an intensive regime of adjuvant chemotherapy was initiated shortly after surgery.It consisted of six courses of VIDE(vincristine, ifosfamide,doxorubicin,etoposide)applied 3-weekly.Most recent restaging 12 months after surgery and after completion of the VIDE-regime showed no signs of residual disease or recurrence.Currently further chemotherapy is being administered with an alternating VAI-(vincristine, actinomycin,ifosfamide)and VAC-regime(vincristine, actinomycin,cyclophosphamide).Continuous remission provided,a second surgery for definitive orthotopic or cutaneous continent urinary diversion is intended upon completion of adjuvant therapy.

Figure 2Upon pathological examination the transrectal biopsy was preliminarily described as a small-blue-round-cell tumor and later classified as a Ewing-like tumor of the periprostatic tissue.(A)Nerve fibre infiltration of the tumor (400×);(B)Tumor formation with close proximity to the prostatic tissue(40×).
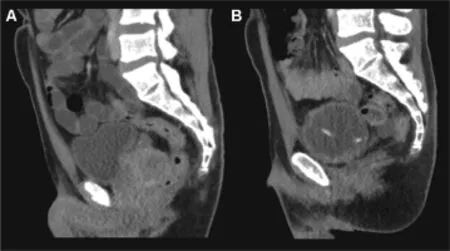
Figure 3Contrast enhanced staging CT-scan of the pelvis(A) and re-staging 2 months after surgery and first cycle of adjuvant chemotherapy(B):no signs of residual disease or recurrence of the Ewing-like tumor.Urine-diversion via cystostomycatheter after closure of the bladderneck.
3.Discussion
Ewing sarcoma is a rare malignant disease in which cancer cells are found in the bone or in soft tissue.Approximately 25%of patients with Ewing sarcoma have metastatic disease at the time of diagnosis.Ewing sarcoma occurs most frequently in teenagers and young adults,with a male/female ratio of 1.6:1[6].In patients aged 10-19 years,the incidence is between nine and 10 cases per million people, the incidence for all ages is even more rare with one case per million people in the United States.The polymorphism (EGR2-gene on 10q21.3)associated with the increased risk is found at a much higher frequency in whites than in blacks or Asians,possibly explaining the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations[7].
For extraosseous primary tumors,the most common primary sites of disease include the following:trunk(32%), extremity(26%),head and neck(18%),retroperitoneum (16%),other sites(9%).The Surveillance,Epidemiology,and End Results(SEER)database was used to compare patients younger than 40 years with Ewing sarcoma who presentedwith skeletal and extraosseous primary sites.Patients with extraosseous Ewing sarcoma were mostly older(mean age of 20 years),white and presented axial primary sites[8]. Extraosseous Ewing sarcoma in adults represent an extremely rare tumor entity with no more than a few hundred reported cases of various forms of presentation in terms of localization and stages of disease[9-11].Overall no definitive causation of this tumor entity has yet been identified[12].
In the presented case we had a 33-year-old Asian male patient with no known exogenic noxae and no known disposition to genetic diseases in the family.
With tumors of the Ewing family general guidelines include neoadjuvant chemotherapy with vincristine,doxorubicin and cyclophosphamide,alternating with ifosfamide and etoposide[13].Furthermore neoadjuvant radiotherapy for improved surgical resection as well as adjuvant irradiation with positive surgical margins are viable options in this disease[14].Definitive radiotherapy is generally considered in those cases deemed unsuitable for surgery, even though the assumed superiority of resection for local control remains a suspect of ongoing studies[15].In our case a solely adjuvant course of regime with initial surgical resection was chosen mostly due to the uncommon initial histopathological presentation of the tumor and therefore an overall inconclusive classification upon preoperative needle-biopsy.As rapid proliferation and highaggressiveness had to be expected prompt surgical extirpation was chosen as primary course of care in order to provide definitive classification and potentially enable targeted systemic treatment of the tumor depending on its entity(e.g.,monoclonal antibody therapy in non-Hodgkin’s lymphoma[16]).
Existing data suggests similar outcome of adult extraosseous Ewing sarcoma compared to that of bone-confined disease in terms of response to multi-modality treatment and the prognostic factors influencing treatment success [17,18].As for the field of urology,individual cases of extraosseous Ewing sarcomas have also been reported in the urinary bladder,kidneys,adrenal glands and even the penis[19-22].To our knowledge only nine other cases of (peri-)prostatic Ewing sarcoma are known worldwide being first described in 2003[23-31].Outcome in these patients was overall very poor with reported survival between as little as 2[24]up to 12 months[30].These survival rates are signi ficantly inferior compared to outcome in the largest available prospective,randomized collection of patients studied today on children or adolescents with extraosseous Ewing sarcomas:130 of 2792 patients(under 21 years of age)registered on three Intergroup Rhabdomyosarcoma Study clinical trials(IRS-I,-II,and-III)from 1972 to 1991 had an extraosseous Ewing sarcoma.Eight-two percent of the patients achieved a complete response with 10-year-overall survival being as high as 62%,61%,and 77%on IRS-I,IRS-II,or IRS-III multi-modal therapeutic protocols, respectively.The therapeutical strategy included mixed groups with adjuvant and neoadjuvant protocols,and no subgroup analyses are available[18].With the lack of larger randomized control trials and the vast majority of studies focusing on osseous Ewing sarcomas in children,choosing the right course of treatment was challenging in this setting.Retrospectively,a neoadjuvant approach would have most likely been the treatment of choice based on current evidence.Also adjuvant radiotherapy after suspicion of positive surgical margins could have been considered.With the patient being in full remission 1 year after diagnosis and a reported average survival of less than 12 months in comparable cases treated with neoadjuvant chemotherapy,our adjuvant approach might be regarded at least non-inferior.Further follow-up could give vital additional information on the optimal treatment sequence in this rare tumor entity.In general transrectal or perineal biopsy seems to be a feasible procedure for the initial diagnosis of unclear pelvic masses.Given the fact that sarcoma is a very rare entity,central registries of rare pelvic tumors would be helpful,in order to get an evidence based,standardized therapy.
4.Conclusion
Sarcomas are a rare but clinically relevant differential diagnosis in pelvic masses of the young adult.Histological confirmation and definitive classification is paramount for optimal treatment,which should include a multimodal approach in an interdisciplinary team.Initial surgery followed by adjuvant chemotherapy might be an alternative approach to neoadjuvant treatment.
Conflicts of interest
The authors declare no conflicts of interest.
[1]Chen QR,Vansant G,Oades K,Pickering M,Wei JS,Song YK, et al.Diagnosis of the small round blue cell tumors using multiplex polymerase chain reaction.J Mol Diagn 2007;9: 80-8.
[2]Rajwanshi A,Srinivas R,Upasana G.Malignant small round cell tumors.J Cytol 2008;26:1-10.
[3]Thway K.Primitive round cell neoplasms.Surg Pathol Clin 2010;4:799-818.
[4]Delattre O,Zucman J,Melot T,Garau XS,Zucker JM, Lenoir GM,et al.The Ewing family of tumors-a subgroup of small-round-cell tumors defined by specific chimeric transcripts.N Engl J Med 1994;331:294-9.
[5]Gu M,Antonescu CR,Guiter G,Huvos AG,Ladanyi M, Zakowski MF.Cytokeratin immunoreactivity in Ewing’s sarcoma:prevalence in 50 cases confirmed by molecular diagnostic studies.Am J Surg Pathol 2000;24:410-6.
[6]Iwamoto Y.Diagnosis and treatment of Ewing’s sarcoma.Jpn J Clin Oncol 2007;37:79-89.
[7]Postel-Vinay S,Ve´ron AS,Tirode F,Pierron G,Reynaud S, Kovar H,et al.Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma.Nat Genet 2012;44:323-7.
[8]Applebaum MA,Worch J,Matthay KK,Goldsby R,Neuhaus J, West DC.Clinical features and outcomes in patients with extraskeletal Ewing sarcoma.Cancer 2011;117:3027-32.
[9]Eyre R,Feltbower RG,James PW,Blakey K,Mubwandarikwa E, Forman D,et al.The epidemiology of bone cancer in 0-39 year olds in northern England,1981-2002.BMC Cancer 2010; 10:357.
[10]Jawad MU,Cheung MC,Min ES,Schneiderbauer MM, Koniaris LG,Scully SP.Ewing sarcoma demonstrates racialdisparities in incidence-related and sex-related differences in outcome.Cancer 2009;115:3526-36.
[11]Esiashvili N,Goodman M,Marcus RB.Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades.J Pediatr Hematol Oncol 2008;30:425-30.
[12]Eyre R,Feltbower RG,Mubwandarikwa E,Eden TOB, McNally RJQ.Epidemiology of bone tumours in children and young adults.Pediatr Blood Cancer 2009;53:941-52.
[13]Grier HE,Krailo MD,Tarbell NJ,Link MP,Fryer CJH, Pritchard DJ,et al.Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone.N Engl J Med 2003;348: 694-701.
[14]Donaldson SS,Torrey M,Link MP,Glicksman A,Gilula L, Laurie F,et al.A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma:end results of POG#8346.Int J Radiat Oncol Biol Phys 1998;42:125-35.
[15]Yock TI,Krailo M,Fryer CJ,Donaldson SS,Miser JS,Chen Z, et al.Local control in pelvic Ewing sarcoma:analysis from INT-0091-a report from the Children’s Oncology Group.J Clin Oncol 2006;24:3838-43.
[16]Marcus R,Hagenbeek A.The therapeutic use of rituximab in non-Hodgkin’s lymphoma.Eur J Haematol Suppl 2007;78: 5-14.
[17]El Weshi A,Allam A,Ajarim D,Al Dayel F,Pant R,Bazarbashi S, et al.Extraskeletal Ewing’s sarcoma family of tumours in adults:analysis of 57 patients from a single institution.Clin Oncol R Coll Radiol 2010;22:374-81.
[18]Raney RB,Asmar L,Newton Jr WA,Bagwell C,Breneman JC, Crist W,et al.Ewing’s sarcoma of soft tissues in childhood:a report from the intergroup Rhabdomyosarcoma Study,1972 to 1991.J Clin Oncol 1997;15:574-82.
[19]Ellinger J,Bastian PJ,Hauser S,Biermann K,Mu¨ller SC. Primitive neuroectodermal tumor:rare,highly aggressive differential diagnosis in urologic malignancies.Urology 2006; 68:257-62.
[20]Okada Y,Kamata S,Akashi T,Kurata M,Nakamura T,Kihara K. Primitive neuroectodermal tumor/Ewing’s sarcoma of the urinary bladder:a case report and its molecular diagnosis.Int J Clin Oncol 2011;16:435-8.
[21]Zhang DW,Jin M,Zhou CJ,Song HC,Ma XL.[Primitive neuroectodermal tumor/Ewing’s sarcoma of the penis in children: a case report and review of the literature].Zhonghua Nan Ke Xue 2012;18:1115-8[Article in Chinese].
[22]Tsang YP,Lang BH,Tam SC,Wong KP.Primitive neuroectodermal adrenal gland tumour.Hong Kong Med J 2014;20: 444-6.
[23]Colecchia M,Dagrada GP,Poliani PL,Messina A,Pilotti S. Primary primitive peripheral neuroectodermal tumor of the prostate.Immunophenotypic and molecular study of a case. Arch Pathol 2003;127:e190-3.
[24]Peyromaure M,Vieillefond A,Boucher E,de Pinieux G, Beuzeboc P,Debre´B,et al.Primitive neuroectodermal tumor of the prostate.J Urol 2003;170:182-3.
[25]Thete N,Rastogi D,Arya S,Singh A,Rao P,Chandge A,et al. Primitive neuroectodermal tumour of the prostate gland: ultrasound and MRI findings.Br J Radiol 2007;80:e180-3.
[26]Kumar V,Khurana N,Rathi AK,Malhotra A,Sharma K, Abhishek A,et al.Primitive neuroectodermal tumor of prostate.Indian J Pathol Microbiol 2008;51:386-8.
[27]Funahashi Y,Yoshino Y,Hattori R.Ewing’s sarcoma/primitive neuroectodermal tumor of the prostate.Int J Urol 2009;16: 769.
[28]Mohsin R,Hashmi A,Mubarak M,Sultan G,Shehzad A, Qayum A,et al.Primitive neuroectodermal tumor/Ewing’s sarcoma in adult uro-oncology:a case series from a developing country.Urol Ann 2011;3:103-7.
[29]Al Haddabi I,Al Bahri M,Burney I.Cytokeratin-positive primitive neuroectodermal tumor of the prostate:case report and review of literature.Indian J Pathol Microbiol 2012;55: 569-71.
[30]Wu T,Jin T,Luo D,Chen L,Li X.Ewing’s sarcoma/primitive neuroectodermal tumour of the prostate a case report and literature review.Can Urol Assoc J 2013;7:458-9.
[31]Shibuya T,Mori K,Sumino Y,Sato F,Mimata H.Rapidly progressive primitive neuroectodermal tumor of the prostate:a case report and review of the literature.Oncol Lett 2015;9: 634-6.
Received 26 August 2015;received in revised form 2 November 2015;accepted 5 November 2015
Available online 19 December 2015
*Corresponding author.
E-mail address:dbarski@lukasneuss.de(D.Barski).
Peer review under responsibility of Second Military Medical University.
http://dx.doi.org/10.1016/j.ajur.2015.11.007
2214-3882/ⓒ2016 Editorial Office of Asian Journal of Urology.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
 Asian Journal of Urology2016年2期
Asian Journal of Urology2016年2期
- Asian Journal of Urology的其它文章
- Prostate specific antigen bounce after intensity-modulated radiation therapy in an Asian population
- Advances in prostate cancer research models:From transgenic mice to tumor xenografting models
- Major histocompatibility complex I upregulation in clear cell renal cell carcinoma is associated with increased survival
- Risk factors for fever and sepsis after percutaneous nephrolithotomy
- Robotic assisted radical prostatectomy accelerates postoperative stress recovery: Final results of a contemporary prospective study assessing pathophysiology of cortisol peri-operative kinetics in prostate cancer surgery
- Glass ampoule in urinary bladder as a foreign body