
再生水补水对河流湿地香蒲根际细菌群落结构影响研究
2016-04-16 07:12黄兴如张琼琼张瑞杰郭逍宇首都师范大学资源环境与旅游学院北京100048北京市城市环境过程与数字模拟重点实验室省部共建国家重点实验室培育基地北京100048
中国环境科学 2016年2期
黄兴如,张琼琼,张瑞杰,郭逍宇*(1.首都师范大学资源环境与旅游学院,北京 100048;2.北京市城市环境过程与数字模拟重点实验室-省部共建国家重点实验室培育基地,北京 100048)
再生水补水对河流湿地香蒲根际细菌群落结构影响研究
黄兴如1,2,张琼琼1,2,张瑞杰1,2,郭逍宇1,2*(1.首都师范大学资源环境与旅游学院,北京 100048;2.北京市城市环境过程与数字模拟重点实验室-省部共建国家重点实验室培育基地,北京 100048)
摘要:以典型的再生水补水河流湿地为例,采用末端限制性片段长度多态性技术(T-RFLP)分析河道不同空间香蒲根际细菌群落结构差异,并借助单因素方差分析(one-way ANOVA)、Spearman等级相关分析和典范对应分析(CCA)方法解析麻峪湿地香蒲根际细菌群落结构空间差异特征的形成原因,以揭示再生水补水过程对河道湿地香蒲根际细菌群落的影响,并尝试找出空间差异形成的驱动因子.结果表明:随再生水干扰强度的增加,各类群细菌群落的丰富度、均匀度及多样性均呈现不同程度的降低趋势;其中γ-变形菌门(Gammaproteobacteria)、δ-变形菌门(Deltaproteobacteria)和绿弯菌门(Chloroflexi)、ε-变形菌门(Epsilonproteobacteria)和放线菌门(Actinobacteria)在再生水影响下均显著降低(P<0.05).Spearman等级相关分析显示pH值、DO(溶解氧)、TDS(总溶解固体)、ORP(氧化还原电位)、Sal(盐度)和NH4+-N(氨氮)与植物根际细菌群落多样性空间演替紧密相关.CCA分析结果进一步表明再生水补水口的上游细菌群落与TN(总氮)、TOC(总有机碳)及重金属Cr、Ni、Cu具有密切关系,这可能与这些污染物累积效应有关;补水口附近植物根际细菌群落则因补水口再生水水质不同而具显著差异,其中第Ⅱa类群主要受到水质变量pH值影响较大,而第Ⅱb类群与T(温度)、ORP和NH4+-N具有较高的相关性;补水口的下游细菌群落则因水体内源杂质及人为活动影响而同样与TOC及持久性痕量重金属生物循环密切相关.
关键词:再生水;T-RFLP;细菌群落多样性;多元统计分析
* 责任作者, 副教授, xiaoyucnu@126.com
作为城市化水平提高的直接负面效应,我国水资源短缺和水体污染日趋严重.作为保护城市水体和拓展水源供给的重要途径,再生水的回用已成为关注的热点[1].据统计到2013年年底,北京市河湖景观用水总量5.7亿m3,其中有3.7亿m3为再生水.但由于再生水水质特性决定了其必然会改变河道生态水文过程和污染物迁移运转,并通过河道垂向渗漏过程影响周边地下水水质特性和污染物迁移运转,进而产生多种生态环境效应[2].基于此,以氨氮为河道水体富营养化主控因素的再生水补水河湖湿地水污染防治成为湿地研究的热点问题之一[3-4].
城市人工湿地不仅具有重要的景观作用,而且具有良好的环境污染修复能力,能有效消除水体氮、磷、各种有机物质、重金属氧化物及病原菌,降低水体生化需氧量(BOD)和总悬浮固体(TSS)含量[5-6],是集观赏、娱乐、污水净化于一体的景观生态环境系统[7],因而在城市景观河道的水质改善中逐渐得到广泛应用[8].人工湿地系统通过基质、水生植物和微生物的物理、化学和生物三重协同作用实现对污水的净化[9-11].其中微生物在水体环境的修复过程中扮演着重要的角色,尤其是植物根际微生物.湿地根际微生物具有丰富的数量和种类,并伴有高效的降解能力[12].一方面,人工湿地中丰富的根际微生物能够有效地降解转化水体中的有机物、氮化合物和磷化合物等污染物[13];另一方面,水生植物通过释放根际分泌物形成“根际效应”,促使根际微生物增强人工湿地的承载能力[14].人工湿地中水力条件、废水特性及各种营养元素的可利用性等条件会直接影响微生物数量、活性及菌群组成等各方面特性[15-16];外源污染物稀释、迁移、转化和降解过程伴随着适应性微生物激活及非适应性微生物抑制的过程[17].因此,湿地植物根际微生物组成及群落结构的变化能够敏感地反映出水体质量,是评价湿地生态系统健康状况的重要指示因子.湿地环境中的微生物多样性是整个系统正常运行的关键[18],然而,目前关于再生水河湖景观补水的相关研究,尤其是人工湿地回用方面多集中于分析人工湿地对再生水中污染物质的去除效率及其净化状况评估[19-20],再生水补水对水生植物群落影响等[21-22],而关于再生水补水对湿地植物根际微生物群落影响鲜有研究.
末端限制性片段长度多态性分析技术(Terminal Restriction Fragment Length Polymorphism, T-RFLP)是基于RFLP技术和荧光标记技术发展起来的[23],依据酶切产生的大量酶切片段和传统的多样性指数相结合可以快速检测和评价由环境扰动而产生微生物群落多样性的变化[24-25].近年来已有学者初步尝试借用统计学方法在海量数据中挖掘数据的内在规律方面的优势对T-RFLP分析中酶切片段进行分析,均取得显著成果[26-28].本文尝试通过基于单因素方差分析(one-way ANOVA)、Spearman等级相关分析和典范对应分析(CCA)相结合的方法对河道不同空间香蒲根际细菌群落结构差异及其驱动环境因子进行分析,研究预期在多元统计分析与T-RFLP相结合的微生物生态学问题分析中进行有益尝试,同时揭示再生水补水对城市河流湿地植物根际细菌群落结构的影响.
1 材料与方法
1.1 研究区概况
以北京市永定河城市景观再生水补水段(上游至三家店,下游至莲石西路)为研究区,该区段地处北京市城区西部,全长约10km,于2011年建成.该研究区位于欧亚大陆东部中纬度地带(116°5'E~116°10'E, 39°53'N~39°57'N),处在东部湿润区和西部干燥区之间,大陆性气候明显.整个流域平均年降雨量约为556~560mm,区域降雨多集中在6~9月.研究区段两岸主要设有两个再生水补水口(中门寺沟补水口和高井排洪渠补水口),年补水总量约200 万m3.研究区内水生植物主要包括香蒲、芦苇、水葱、浮萍等,植被覆盖率可达70%~90%,其中香蒲最为普遍.
1.2 样品采集及理化性质分析
本研究分析的样品于2012年9月采自门头沟区永定河段麻峪湿地上游至下游典型的城市河段,实验样品为研究区内城市河道香蒲根际新鲜土壤样品.依据《永定河生态功能区划》合理布点采样,其中X1和X2采样断面位于再生水补水口上游河段,X3、X4和X5采样断面则分别设在中门寺沟和高井排洪渠补水口附近,X6、X7 和X8采样断面选定在补水口下游约2000m处,详见图1.
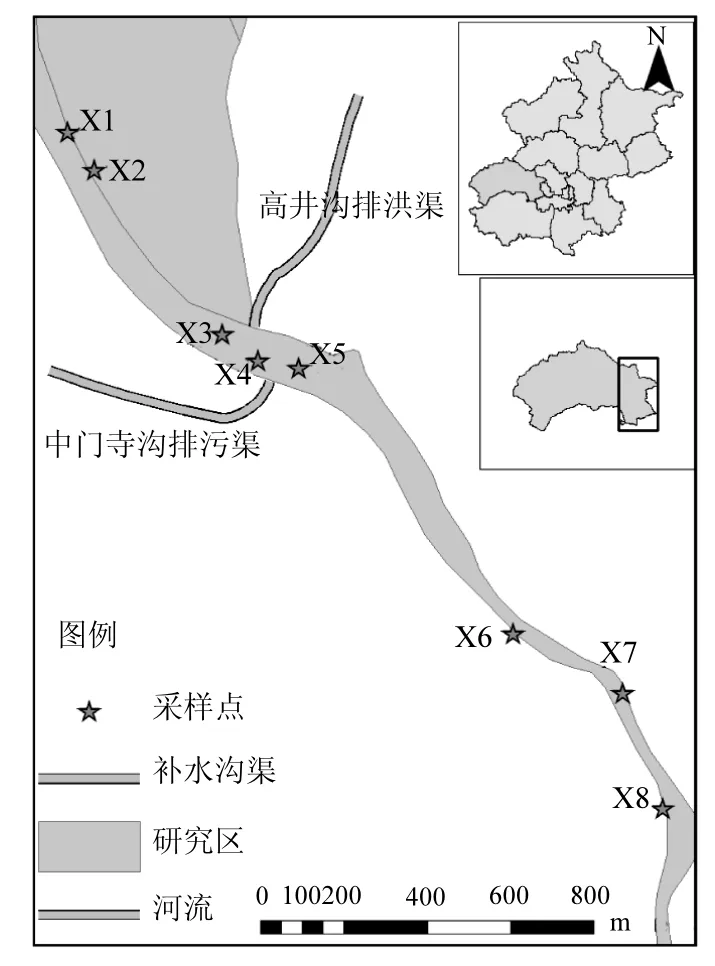
图1 研究区采样点位置示意Fig.1 Location and distribution of the sampling points in study area
每一断面从中间至两岸湿地植物根际均匀采样,混合均匀后置于无菌密封袋中保存,冷藏带回实验室处理.同时用Hydrolab Datasonde5 5X水质仪进行现场水质监测,包括T (℃)、pH值、氧化还原电位(ORP)、盐度(Sal)、总溶解性固体(TDS)、溶解氧(DO)等参数.带回实验室样品分两部分处理,其中一部分进行常规理化指标分析,其中总氮(TN)、总磷(TP)、总有机碳(TOC)均采用国标法测定,NH4+-N采用2mol/L KCl浸提—靛酚蓝比色法测定,重金属采用原子吸收光谱法来测定.剩余根际土壤样品于-20℃下保存,用于微生物群落结构分析.各样点理化性质测定结果见表1.
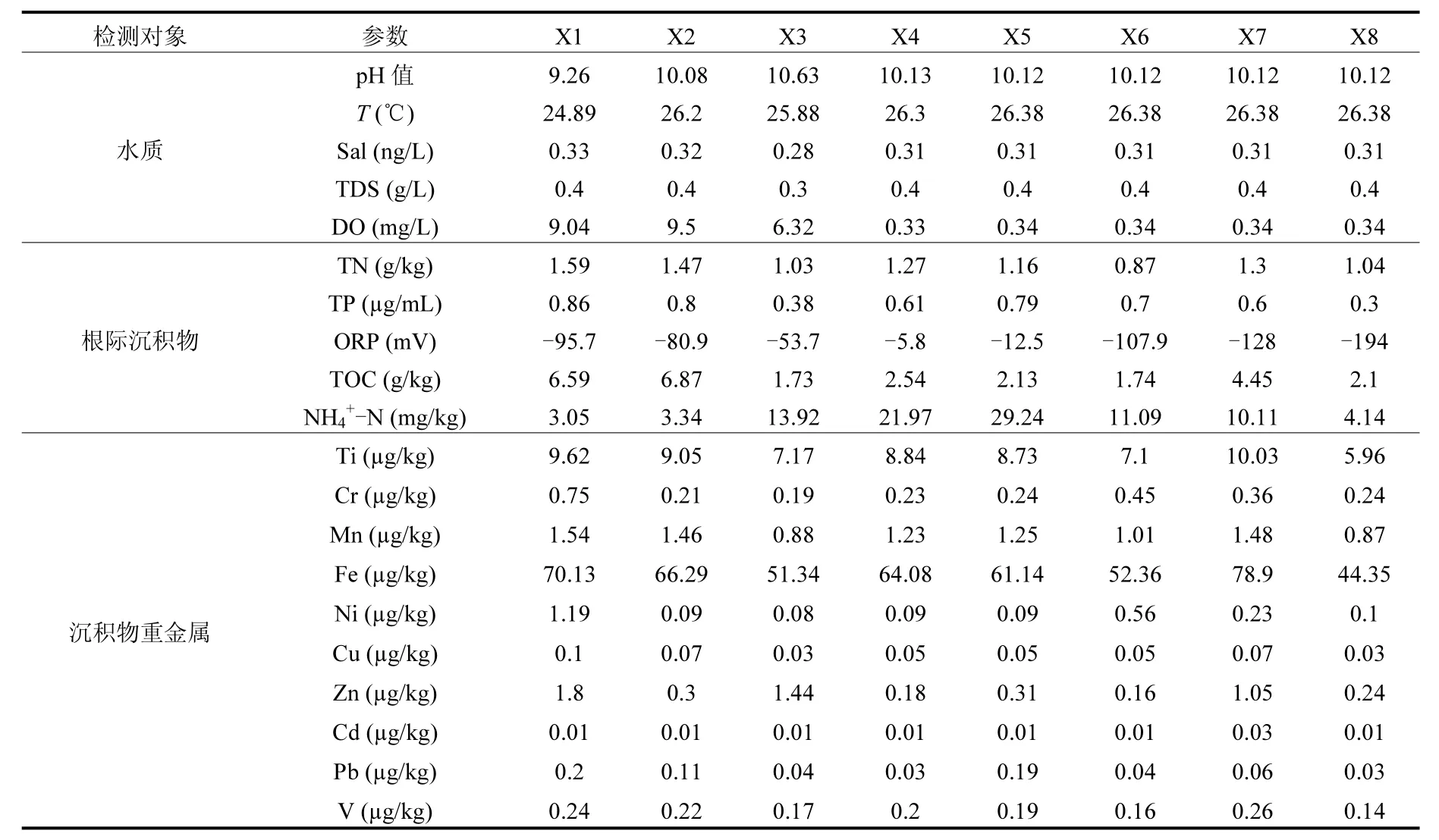
表1 水质及香蒲根际沉积物理化性质Table 1 Water quality and physicochemical properties of cattail rhizosphere sediment
1.3 基于T-RFLP的微生物群落多样性分析
1.3.1 DNA提取 采用PowerSoil DNA IsolationKit 12888-50(MOBIO提供)提取植物根际沉积物样品细菌总DNA,操作步骤按照使用说明书进行.提取总DNA经0.8%(m/V)琼脂糖凝胶电泳分离鉴定,得到的DNA样品放置于-20 ℃温度条件下保存、备用.
1.3.2 细菌16S rDNA的PCR扩增 用5'端经6-FAM修饰的引物27f(5'-AGAGTTTGATCCTGGCTCAG-3')和无修饰的1492r(5'-GGTTACCTTG TTACGACTT-3')对总细菌16S rDNA进行PCR扩增.25µL扩增体系包含2µL DNA 模板,12.5µL 2×Taq PCR Master Mix,1µL 10μmol/L 27f 和10μmol/L 1492r,8.5µL ddH2O.PCR扩增程序设置条件为:95℃预变性5min;95℃变性50s, 55℃退火50s,72℃延伸1min,30个循环;最后72℃延伸7min,4℃保存.荧光PCR产物采用0.8%的琼脂糖凝胶电泳检测后,用锡纸密封包裹避光置于4℃保存,以备酶切消化.
1.3.3 末端限制性片段长度多态性(T-RFLP)分析 分别采用限制性内切酶MspⅠ、HhaⅠ和RsaⅠ对16S rDNA PCR产物酶切.酶切反应体系(10μL): PCR产物5µL,MspⅠ/HhaⅠ/Rsa Ⅰ0.5µL, 10× buffer 1µL,ddH2O 3.5µL,将体系混匀后,置于恒温培养箱37℃反应4h.在65℃条件下水浴15min使限制性内切酶失活,终止消化反应.随后将酶切产物送至天根生物工程有限公司进行基因扫描(GeneScan),获得TRFLP图谱.
1.3.4 数据处理与分析 (1) T-RFLP数据预处理 T-RFLP谱图用Peak Scanner进行分析.舍去小于50bp和大于500bp的片段.对于细菌,由于相对数量过小的限制性末端片段(T-RFs)不会对群落的特性产生明显的影响[29-31],故在本分析中舍去了相对数量<1%的T-RFs,然后分别计算图谱中每一个峰的峰面积与所有峰总面积的比值,最终形成8个样品的224个T-RFs类型的相对峰面积组成的原始数据矩阵.
(2) 分类与排序 将每个T-RF所占的百分比作为权重导入Primer软件,聚类方法选择组间平均距离法,距离选择平方欧氏距离,做出聚类分析图,同时进行MDS排序.
(3) 细菌群落多样性分析 每一个T-RF类型至少代表一种细菌类群,以各T-RF类型的丰富度及其对应的相对丰度计算细菌群落物种的香侬指数(Shannon index H′)、辛普森指数(Simpson index 1/D)和均匀度指数(Eveness index J′),分析湿地植物根际细菌多样性的空间差异.其中
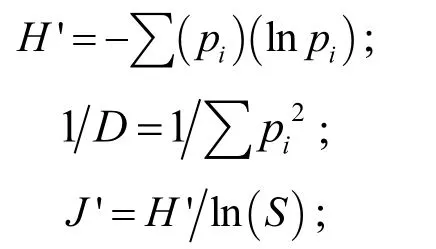
式中:pi代表片段的相对丰度;D是辛普森优势度指数,其与多样性成反比;S是T-RFs的总数,可用以表示物种丰度( richness).
(4) 基于传统T-RFs片段与多元统计相结合分析 将MspⅠ、HhaⅠ和RsaⅠ3种限制性内切酶消化的T-RFLP图谱属性数据上传到Phylogenetic Assignment Tool (PAT, https://secure. limnology.wisc. edu/trflp/newuser.jsp)网站,并结合网站MiCA (http://mica.ibest.uidaho.edu/pat. php)通过Virtual Digest (ISPaR)模块产生的基础数据库对起主要作用T-RFs类型的系统发育分类进行推测.之后应用单因素方差分析、Spearman等级相关分析、典范对应分析(CCA)等多种统计分析方法对河道不同空间香蒲根际细菌群落结构差异及其驱动环境因子进行分析研究.各类统计方法由Office Excel 2007和SPSS18.0实现.
2 结果与讨论
2.1 不同内切酶消化多样性比较
表2为8个样品经不同限制性内切酶MspⅠ、HhaⅠ和RsaⅠ消化所获得的T-RFLP图谱文件信息.同一个样品经MspⅠ、HhaⅠ和RsaⅠ消化后,反映出的总T-RFs数和总峰面积具一定的差异.经比对发现,MspⅠ和HhaⅠ消化后的T-RFs多样性明显优于RsaⅠ,即MspⅠ和HhaⅠ酶切结果能够揭示更高的丰富度.基于此,后续的多元统计分析均基于MspⅠ和HhaⅠ消化结果进行分析.
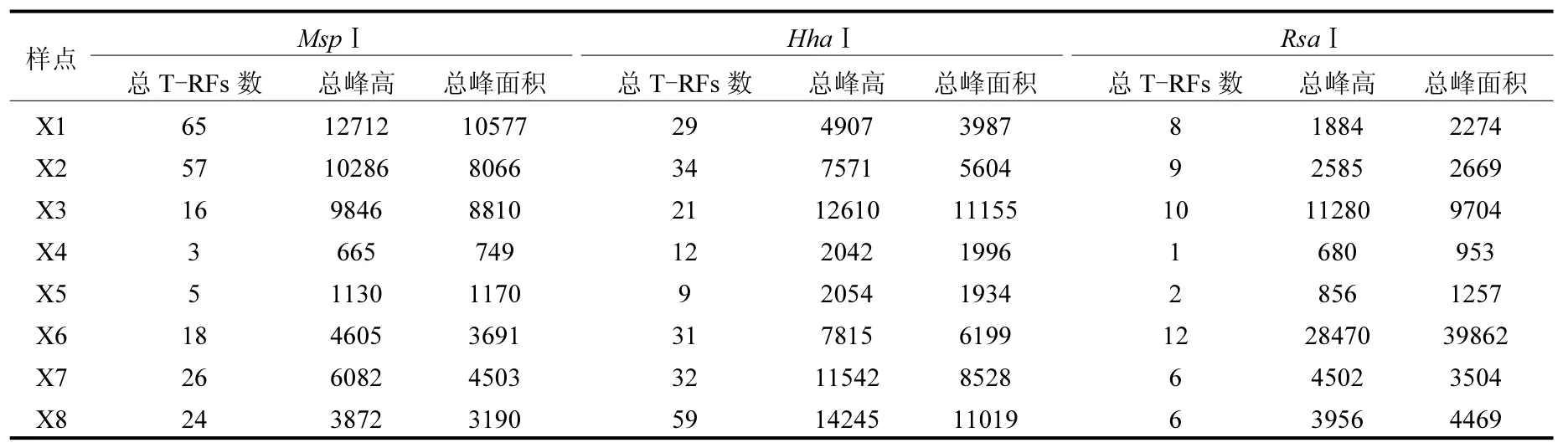
表2 香蒲根际样品细菌经MspⅠ、HhaⅠ和RsaⅠ限制性内切酶消化结果总汇Table 2 Data summery of cattail rhizosphere bacterial samples with MspⅠ、HhaⅠand RsaⅠdigestion
2.2 基于T-RFLP图谱的聚类及MDS排序分析
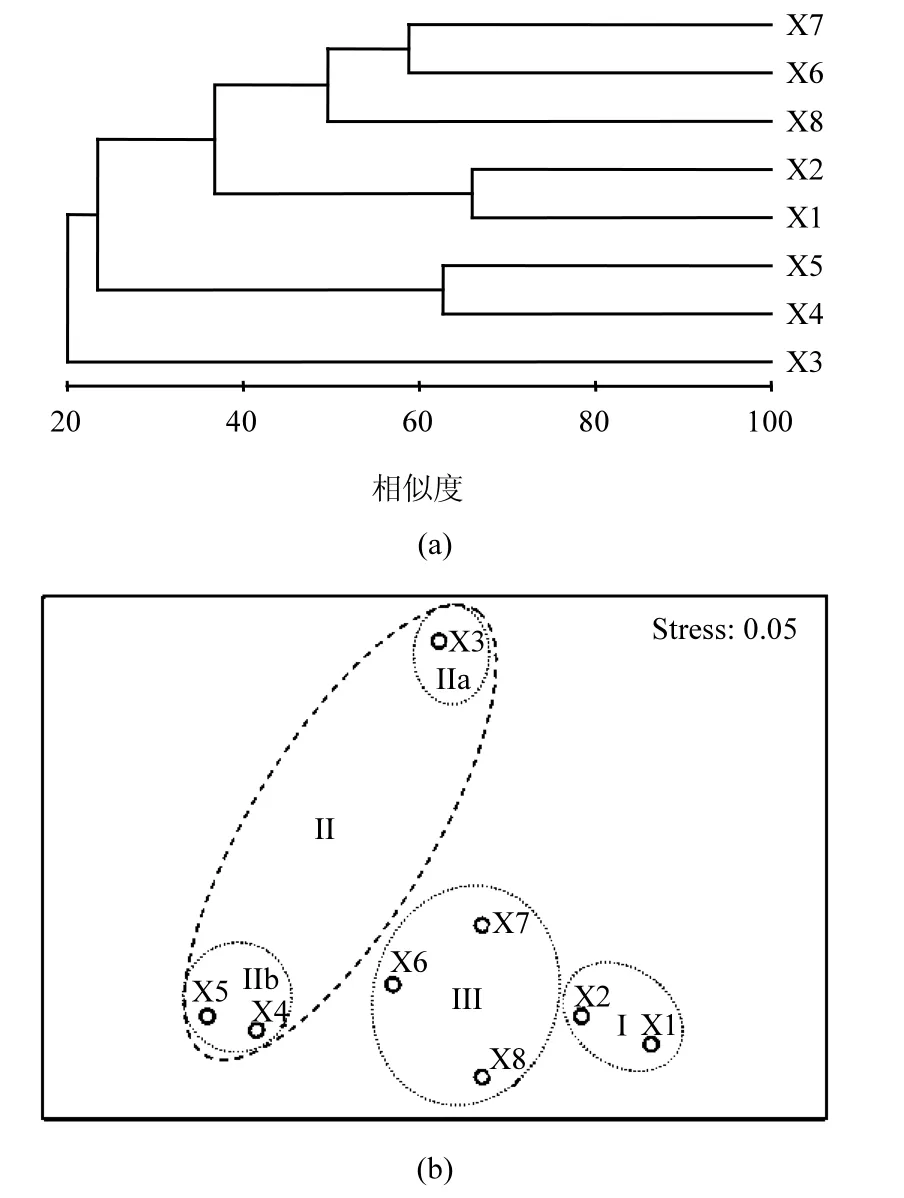
图2 基于T-RFLP图谱的香蒲根际细菌群落结构聚类分析及MDS排序Fig.2 Dendrogram of hierarchical cluster analysis and MDS ordination of cattail rhizosphere bacteria based on the T-RFLP profiles
聚类分析能够反映样品间细菌群落的相似性及差异性.如图2所示,细菌群落结构在再生水河道补水口和上下游均发生较明显的变化.以聚类相似性50%为标准,并结合排序结果将所采集的样点划分为四大类:第Ⅰ类由再生水河道补水口上游X1与X2样点组成,再生水补水口附近X3样点为第Ⅱa类,X4和X5样点为第Ⅱb类,而第Ⅲ类则包括再生水补水口下游约2000m外的X6、X7和X8样点.综合分类和排序结果可以看出,随再生水干扰程度减弱,各类群样点沿排序图左上角至右下角呈现明显的变化规律.类Ⅱ样点根际细菌群落结构与其他各样点间相似性存在较大差异,说明补水口附近底泥的生物物理化学过程显著区别于主河道底泥,需进一步结合微生物组成成分解析;其中排污口附近样点类Ⅱa和类Ⅱb因不同补水口再生水水质差异影响而在排序图中具较广分布格局,第Ⅲ类样点随距补水口距离远近差异同样较第Ⅰ类样点具相对较广分布格局,同时发现类Ⅲ与类Ⅰ具有相对较高的相似性.
2.3 基于单一酶切片段多样性表征参数变异特征
根据MspⅠ、HhaⅠ和RsaⅠ酶切结果,计算了不同酶切样品中各片段的丰度,并定义T-RFs片段丰度值>4%的类型为优势菌群,而片段丰度值<1%的类型为偶见菌群,其余为非优势菌群[32],采用多样性指数、菌群丰度和菌群T-RFs片段数表征各样点细菌群落多样性特征,具体见表3、表4. 3种酶切结果均显示随再生水干扰强度的增加细菌群落各多样性指数均出现不同程度的降低趋势.再生水补水口附近样点(X3、X4、X5)因其受补水水质影响,具有最高优势菌群丰度和最低优势菌群片段数,同时具有相对较低的偶见菌群丰度和偶见菌群片段数,二者综合作用导致其细菌群落具最低均匀度和丰富度.而位于再生水补水口上游的样点(X1、X2)因其细菌群落具最低优势菌群丰度和相对较高的优势菌群片段数,因而群落具最高均匀度和丰富度.不同酶切类型相比较,HhaⅠ和MspⅠ酶切片段的丰富度及其多样性指数明显优于基于RsaⅠ酶切的多样性.不同菌群类型相比较,不同环境条件下优势菌群基于不同酶切类型间变异规律更为一致,而偶见菌群则受酶切类型的影响规律各异.
2.4 细菌群落多样性与环境因子的相关分析
通过单因素方差分析对表1中各理化性质指标进行差异性检验,结果表明理化指标ORP、TOC 和NH4+-N在不同分类样点间具显著差异(P<0.05),而Sal、TDS和DO则呈极显著差异(P<0.01).由表1可见,理化指标Sal和TDS在补水口附近显著变化,而后趋于稳定;而DO显著降低的同时,ORP缓慢增加,而后又在样点X4处急剧下降,这可能是由于再生水补水引起底泥扰动,从而促使根际土壤中ORP略微增加.除此之外,NH4+-N在补水口附近显著增加,而在下游明显降低,可见湿地对氨氮的去除发挥重要作用.相对于其他位点,补水口上游的营养指标TN,TP和TOC最高,这可能是由于该采样点位于三家店水库,缓慢的水流致使有机物和其他物质沉积所致.
为进一步分析湿地净化系统中香蒲根际细菌多样性与环境因子间的响应关系,将植物根际细菌多样性表征参数与环境因子进行Spearman等级相关分析,结果见表5.从表5可看出,pH值、Sal、TDS、DO、ORP和NH4+-N六项指标与植物根际细菌多样性空间演替具有密切关系.研究者们一再证明, pH 值是细菌多样性和群落结构演替的重要决定因子,并且驱动细菌群落的空间分布,其微小波动就可能诱使原始固有优势菌群组成发生改变[33-35].该研究区水质为碱性(pH:9.26~10.63),受再生水补水影响pH值有所上升,升高的pH值可通过影响不同种类细菌的生长状况(包括绝灭、繁衍、种的形成等),直接影响多样性;也可通过影响湖泊生态系统中的其他环境因子(如有机物质的分子结构)来间接影响水体细菌群落的结构和多样性[36-38].微生物的呼吸作用和发酵过程是有机污染物的重要去除机制,其去除过程依赖于ORP,宽泛的ORP范围有利于多种污染物的去除[11];而DO是改善湿地氧化还原环境的重要环境因子,是微生物群落演替的关键因子[39];由此可见,在再生水补水的影响下,DO及ORP的降低易致使细菌群落特征发生改变.诸多研究表明盐度同样是影响细菌群落组成和多样性的一个重要因素,在细菌群落演替中扮演重要角色[40-41];且发现细菌丰度在不同盐度水体呈生态学上经典的“单峰模型”,即在相同营养水平下,细菌在中等盐度的水体中拥有最多的生态位,细菌丰度最高[42-43].这也就映证了在该研究中,低盐度淡水生态系统湿地植物根际细菌的丰度及多样性均与水体的盐度显著正相关.Wu等[43]通过研究位于青藏高原青海–西藏段上16个高山湖泊,发现盐度是控制浮游细菌丰度与群落组成的主导环境因子,盐度含量高的湖泊,细菌的丰度也高;湖泊水体细菌多样性随着水体盐度增加并未呈现减少趋势.这一结论与该研究区湿地植物根际细菌分布规律相一致.氨氮与细菌群落结构组成呈现最大相关性,是微生物群落结构变化的主要影响因子[44];尤其是水中厌氧氨氧化细菌的丰度和多样性与NH4+-N含量具显著相关性[45].
2.5 基于CCA排序的群落结构变异的环境解释
利用MspⅠ酶切的8个样点和环境因子进行趋势对应分析(CCA),进一步分析再生水补水湿地净化系统中香蒲根际细菌群落结构特征及其成因.排序结果表明,CCA 排序图第一轴AX1和第二轴AX2的特征值累计占总特征值的66.9%,排序图包含了大部分的信息,排序效果良好,结果见图3(a).该研究区的8个样点在CCA轴上得到了很好的分布,总的来看,与第一排序轴相关性高的环境因子是pH值、TDS和Sal,在x轴方向上表现出第Ⅱa类与其他群落类型之间的差异;而与第二排序轴相关性最高的环境因子是NH4+-N,其次是ORP、T、TOC、DO及重金属Ni、Cr、Cu,基于分类的各类型样点在y轴方向上从下到上表现为“类Ⅰ>类Ⅲ>类Ⅱa>类Ⅱb”的变化趋势,这种变化趋势与多样性指数变化趋势一致,同时也与再生水补水干扰强弱相呼应.类Ⅱa和Ⅱb分别独立于其他类群位于图最上方和最右方,表明其具独特的微生物生态特征,其中类Ⅱa受环境因子pH影响较大,而类Ⅱb与T、ORP、 NH4+-N具有较高的相关性,即第Ⅱb类群与氨氮生物循环具密切关系;环境因子ORP、NH4+-N、TOC、DO以及重金属Cr、Ni、Cu对类Ⅰ和类Ⅲ群落空间分布贡献较大,其中ORP、NH4+-N表现负效应,TOC、DO以及重金属Cr、Ni、Cu表现正效应,表明第Ⅰ类群和第Ⅲ类群群落与TOC和持久性痕量重金属生物循环密切相关.由图3(b)可知,以MspⅠ酶切CCA聚类图和以HhaⅠ酶切CCA聚类图具有相似的结果.
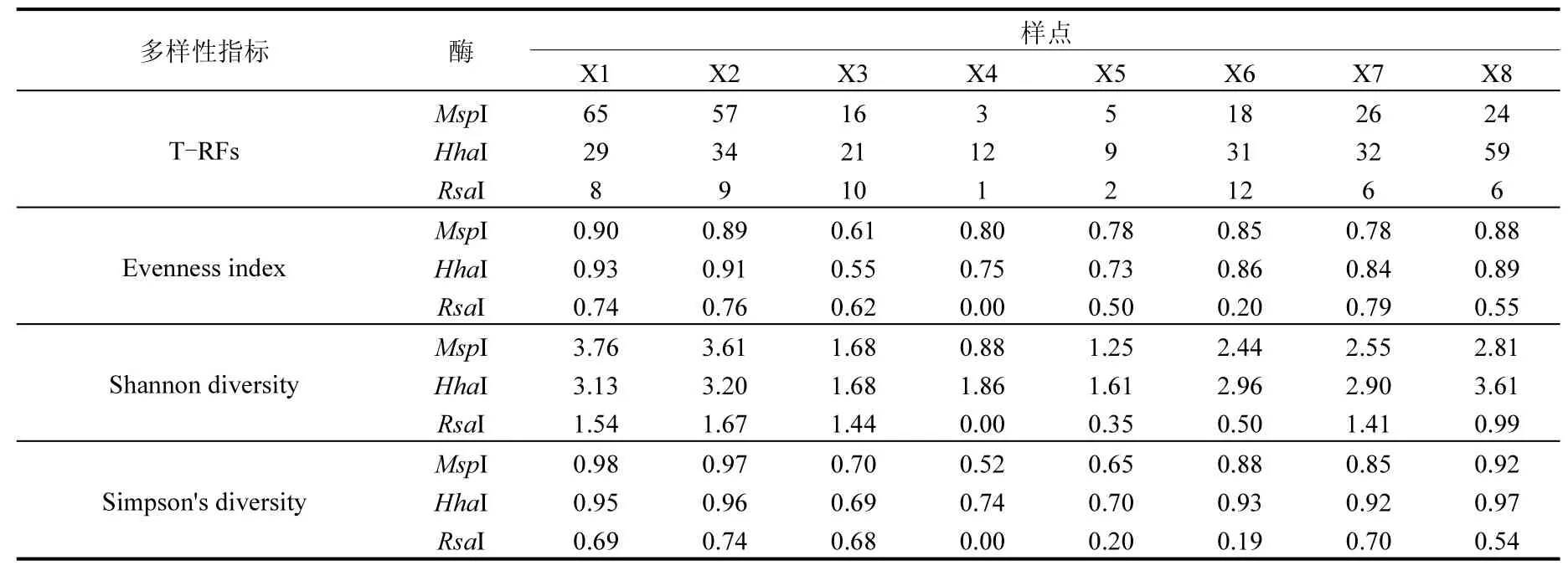
表3 细菌群落多样性分析Table 3 Diversity analysis of cattail rhizosphere bacterial community
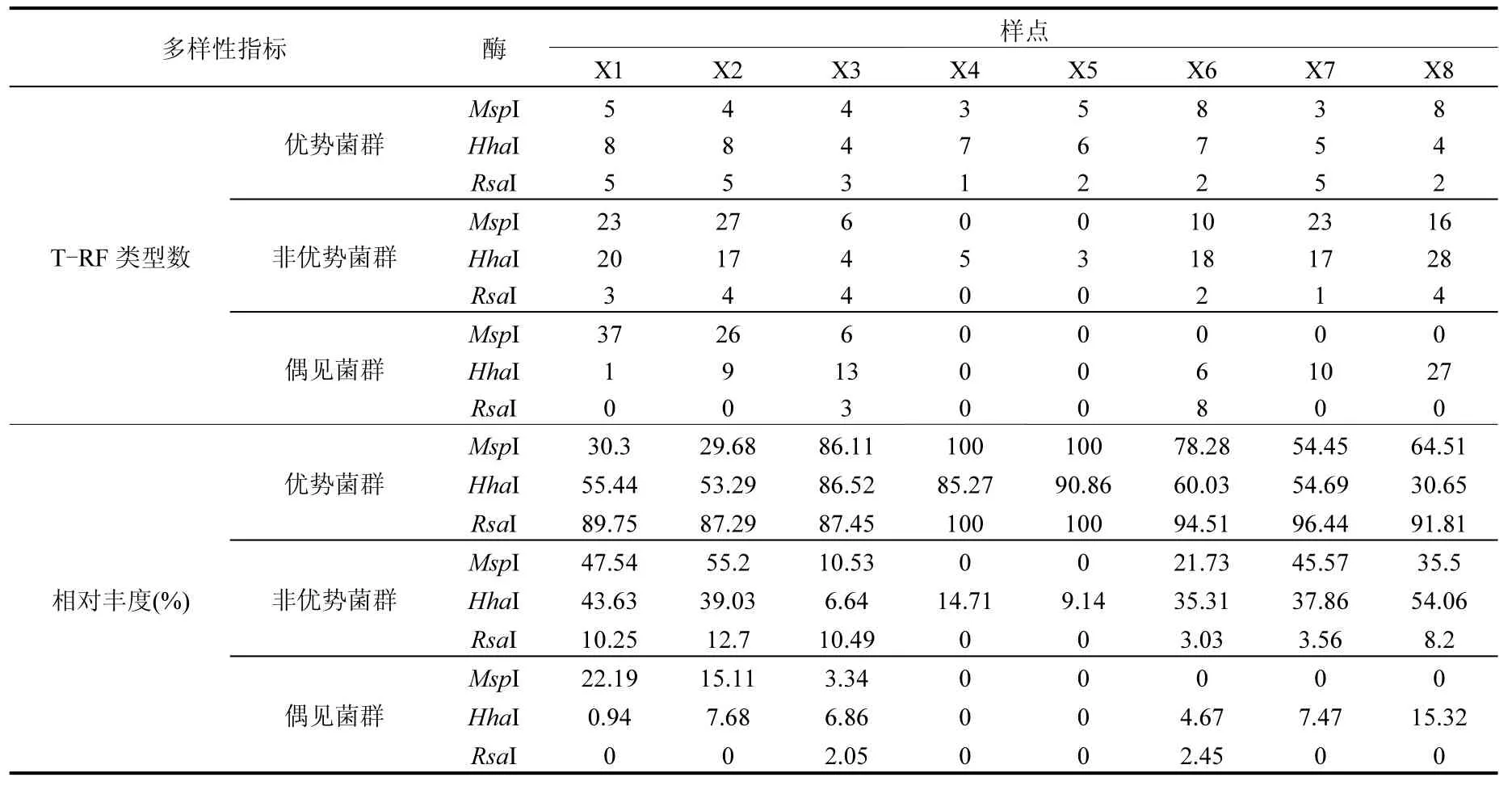
表4 细菌群落结构特征分析Table 4 Structural characters of cattail rhizosphere bacterial community
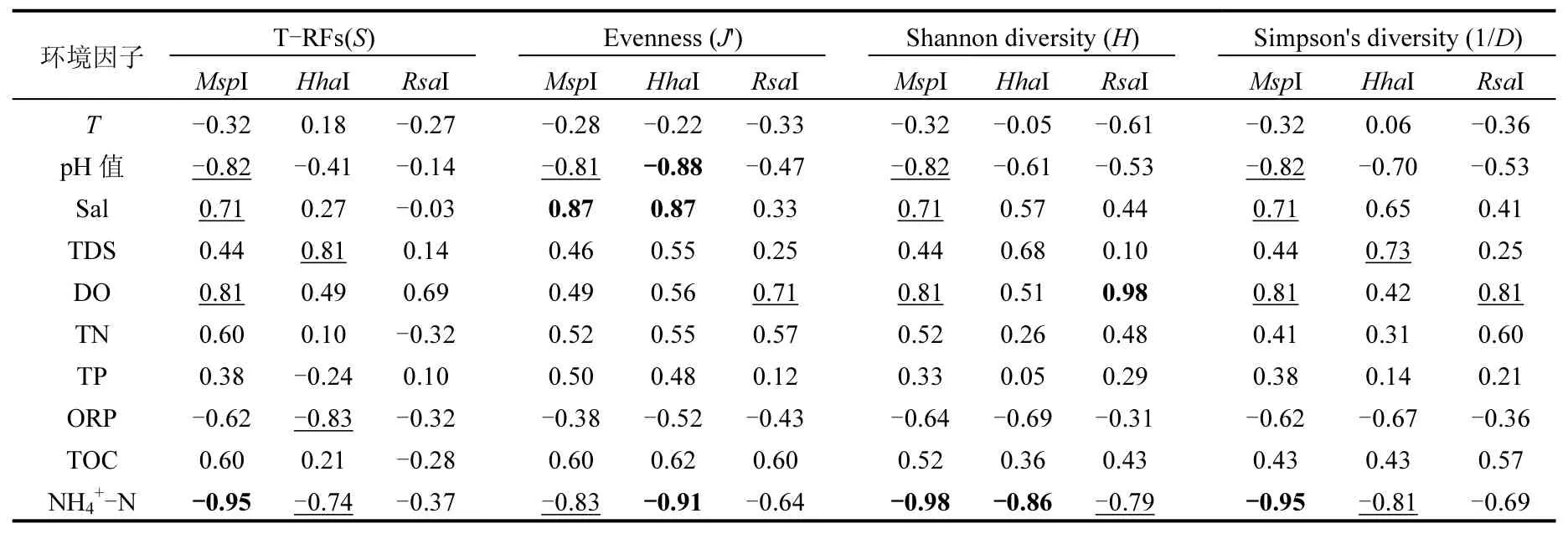
表5 香蒲根际细菌特性指标与环境因子的相关性Table 5 Relationship between the bacteria characteristic parameters and the wetland chemical properties using Spearman’s correlation analysis
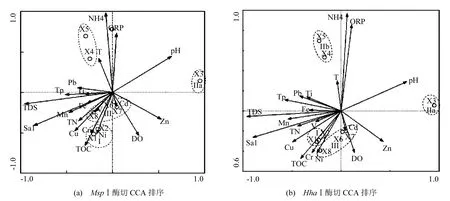
图3 再生水补水河道植物根际采样点与环境因子的CCA排序Fig.3 CCA sequence diagram of the sample point and environmental factors on Reclaimed water in river plant rhizosphere
2.6 基于Mica对比的群落结构分析
2.6.1 优势菌群分析 通过Virtual Digest (ISPAR)模块产生的基础数据库对起主要作用T-RFs类型的系统发育分类进行推测,其中有将近20%暂不能确定,显示为非培养.另外,有个别的T-RFs类型在数据中无匹配.之后通过计算其细菌T-RFs的比例,以占据整个T-RF的4%以上为优势菌种,筛选结果见表6.
依据T-RFLP片段的Mica比对结果,试图通过优势菌群结构特征来反映湿地植物根际细菌群落空间差异.由表6可知,反应再生水典型特征的第Ⅱb类群优势菌属包含热袍菌属(Thermotoga sp.),即说明类Ⅱb细菌群落受温度影响较大,同时CCA排序分析也具有相似结论.仅出现在第Ⅲ类的优势菌属为Bacillus sp.和Lautropia sp.,有报道称Bacillus sp.可迅速降解包括鱼的排泄物、残余饲料、浮游藻类尸体和池底淤泥在内的有机物,使之生成硝酸盐、磷酸盐、硫酸盐等无机盐类,从而降低水中COD、BOD的含量,维持良好的水域生态环境[46];而Lautropia sp.常分布于人体口腔内[47],据此可初步推测该样点细菌群落受水体内源杂质及人为活动影响较大.Flavobacterium sp.为类Ⅰ和类Ⅲ共有的优势菌属,据文献可知,其常聚集于富含硝酸盐的富营养水体中,可有效降低水体中氮的含量,并能够分解代谢水体中的有机物质[41,48-49],长期存在于该净化系统中,对净化系统的生态环境稳定性起重要作用.除此之外,第Ⅰ类优势菌属还包括Pseudomonas sp.、Geitlerinema sp.、Sulfurospirillum sp.、Delftia sp.和Acidovorax sp.;Pseudomonas sp.是一种有机污染中普遍存在的菌属,可以利用包括单碳在内的许多有机物作为自身的能量和碳源,以有机氮或无机氮为氮源进行化能营养生活[50];而Geitlerinema sp.是专性属于底栖生物环境,隶属于蓝菌门(Cyanophyta)的颤藻目(Oscillatoriales),与水环境中氮的循环具有密切关系已得到普遍的认同[51]; Sulfurospirillum sp.属于异养反硝化细菌,可有效降解硫酸盐和硝酸盐[52];而Delftia sp.可降解苯胺,塑化剂以及六价铬[53];由此可见,第Ⅰ类细菌群落受TN、TOC及重金属Cr影响较大,这与CCA排序分析结果较为一致.
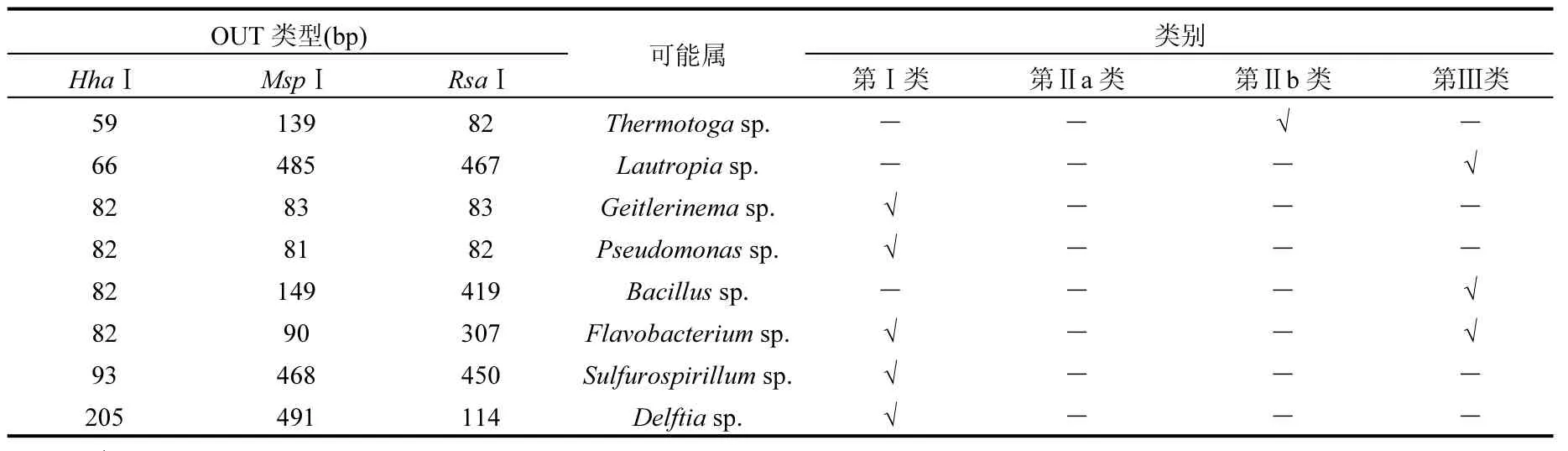
表6 不同类群中植物根际细菌群落可能优势属Table 6 Possible dominant genera of plant rhizosphere bacteria community based on different classification
2.6.2 基于多酶切综合比对细菌群落多样性变异分析 为进一步分析基于门水平的不同样点间植物根际细菌群落多样性变异特征,利用PAT多酶切比对结果,依据属数量分析各类细菌群落门水平的多样性,并进一步通过单因素方差分析筛选出各类群落间多样性具有显著差异的门,结果见图4.由图4可看出随再生水干扰强度的增加,各位点属数量呈现出“类Ⅰ>类Ⅲ>类Ⅱa>类Ⅱb”的变化趋势.这与基于单一酶切片段多样性变异特征分析结果相符,即再生水补水口的上游细菌群落多样性与下游细菌群落趋于相似,而显著区别于补水口附近样点.说明再生水补水直接影响到麻峪湿地微生物群落结构的变化以及微生物群落组成,使得补水口附近细菌群落多样性显著降低;而下游相对升高的细菌群落多样性则可能是由于受再生水补水影响的细菌群落在下游得以逐渐恢复,亦或是因为对逆境产生抗性的一种表现.
单因素方差分析结果表明,多样性在各类群落间具有显著差异的γ-变形菌门(Gammaproteobacteria)、δ-变形菌门(Deltaproteobacteria),绿弯菌门(Chloroflexi), ε-变形菌门(Epsilonproteobacteria)和放线菌门(Actinobacteria)主要在再生水补水口的上游富集,下游次之,补水口附近最少,这种分布特征除与再生水水质特征相关外,还可能与河流水体本身所含营养物质有关.再生水补水口的上游(X1,X2)因累积效应含有相对较高的营养指标TN,TP和TOC;下游(X6,X7,X8)则因内、外部原因也同样含有大量的有机物.与此同时,研究发现Gammaproteobacteria多存在于富营养环境[54];Actinobacteria属于淡水水体中的优势菌,倾向于存在静水水体中[41],且有机物、氮和磷含量较高的污染水体易产生大量放线菌属的细菌[55];Deltaproteobacteria多数为厌氧硫酸盐还原菌,能够分解多种有机化合物;Chloroflexi属于绿色非硫细菌,常分布于水合物较少而有机质丰富的热液沉积物中,可见上游生境中丰富的有机质为该门细菌的生长提供了底物[56].综合CCA排序分析结果及优势菌群分布特征,解析了湿地香蒲根际细菌群落结构空间差异特征的形成原因.
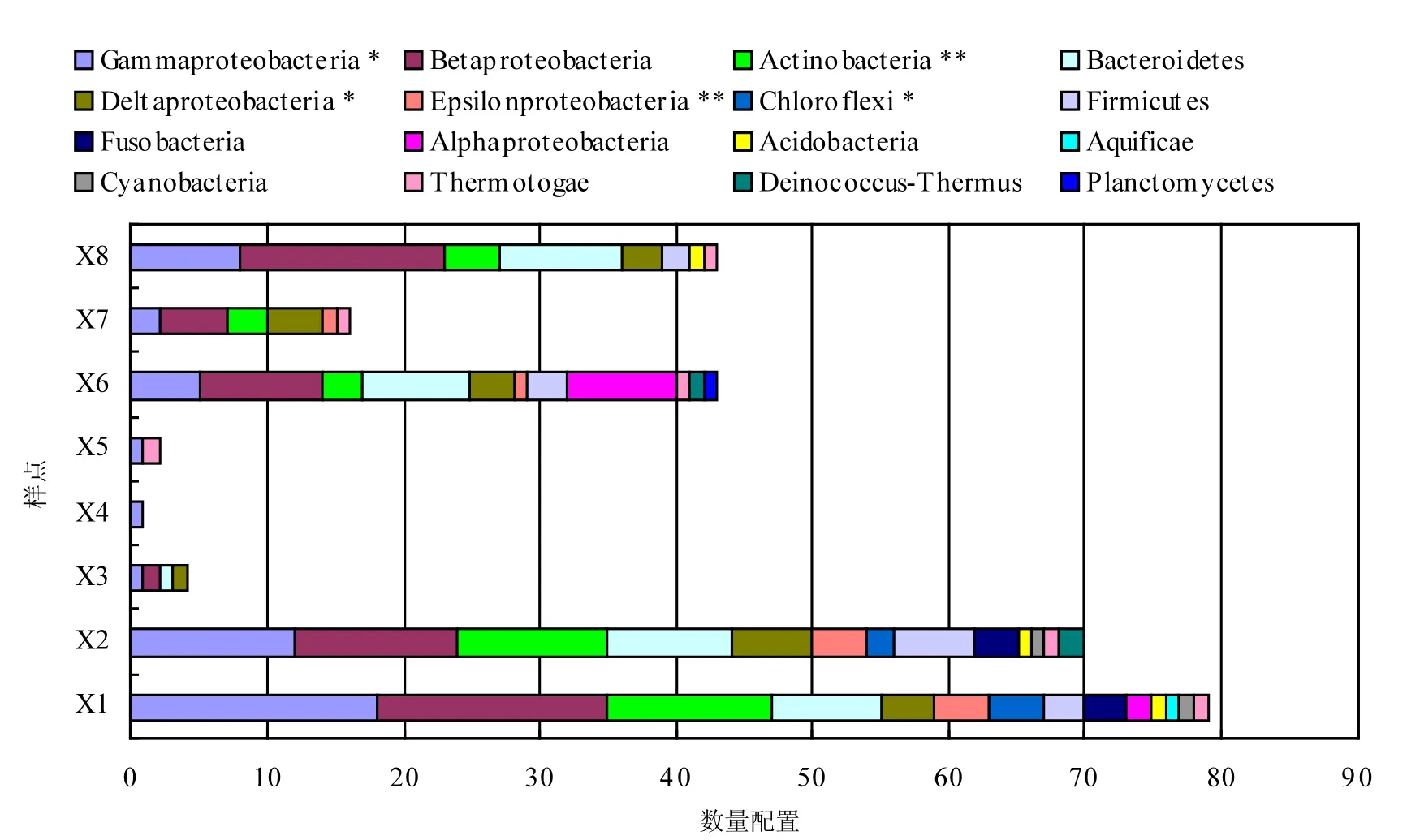
图4 再生水补水影响下基于门水平的香蒲根际细菌群落多样性分类Fig.4 Barchart of bacterial community diversity in cattail rhizosphere classification based on phylum level “**”和“*”分别表示再生水补水影响下基于门类水平的同一菌门在不同类群中的极显著差异分布和显著差异分布
3 结论
3.1 污水处理厂的再生水直接影响到麻峪湿地微生物群落结构的变化以及微生物群落组成,湿地植物根际细菌群落多样性随着再生水干扰强度的增加呈下降趋势“类Ⅰ>类Ⅲ>类Ⅱa>类Ⅱb”,即再生水补水口的上游细菌群落多样性与下游细菌群落趋于相似,而显著区别于补水口附近样点.
3.2 细菌群落多样性与环境因子相关分析表明,水质变量pH、DO、TDS、ORP、Sal和NH4+-N六项指标与植物根际细菌多样性空间演替具有密切的关系.
3.3 再生水补水口的上游细菌群落(X1,X2)与 TN、TOC及重金属Cr、Ni、Cu具有密切关系, 这可能与这些污染物累积效应有关;补水口附近植物根际细菌群落(X3,X4,X5)则因补水口再生水水质不同而具显著差异,其中第Ⅱa类群受水质变量pH影响较大,而第Ⅱb类群与T、ORP和NH4+-N具较高的相关性;补水口下游细菌群落(X6,X7,X8)则因水体内源杂质及人为活动影响而同样与TOC及持久性痕量重金属生物循环密切相关.
参考文献:
[1] Crook J, Surampalli R Y. Water reclamation and reuse criteria in the US [J]. Water Science and Technology, 1996,33(10):451-462.
[2] 刘 克.北京市典型河湖再生水补水生态环境效应研究 [D].北京:首都师范大学, 2012.
[3] 李 兵,林炜铁.一株好氧反硝化芽孢杆菌的脱氮特性研究 [J].水生态学杂志, 2009,2(3):48-51.
[4] 曾 薇,李 磊,杨莹莹,等.A2O工艺处理生活污水短程硝化反硝化的研究 [J]. 中国环境科学, 2010,30(5):625-632.
[5] Zhao S M, Hu N, Chen Z J, et al. Bioremediation of reclaimed wastewater used as landscape water by using the denitrifying bacterium Bacillus cereus [J]. Bulletin of Environmental Contamination and Toxicology, 2009,83(3):337-340.
[6] Thurston J A, Gerba C P, Foster K E, et al. Fate of indicator microorganisms, giardia and cryptosporidium in subsurface flow constructed wetlands [J]. Water Research, 2001,35(6):1547-1551.
[7] 熊 薇.城市湿地植物根际细菌群落多样性时空变异及其水环境解释 [D]. 北京:首都师范大学, 2013.
[8] Cui F, Yuan B, Wang Y. Constructed Wetland as an Alternative Solution to Maintain Urban Landscape Lake Water Quality: Trial of Xing-Qing Lake in Xi’an City [J]. Procedia Environmental Sciences, 2011,10:2525-2532.
[9] Malecki-Brown L M, White J R, Reddy K R. Soil Biogeochemical Characteristics Influenced by Alum Application in a Municipal Wastewater Treatment Wetland [J]. Journal of Environmental Quality, 2007,36(6):1904-13.
[10] Sundberg C, Stendahl J S K, Tonderski K, et al. Overland flow systems for treatment of landfill leachates: Potential nitrification and structure of the ammonia-oxidising bacterial community during a growing season [J]. Soil Biology and Biochemistry, 2007, 39(1):127-138.
[11] 籍国东,倪晋仁.人工湿地废水生态处理系统的作用机制 [J].环境污染治理技术与设备, 2004,5(6):71-75.
[12] Feng C L, Li K L, Li Y. Community characteristics and purification mechanism of microbial in wetland [J]. Journal of Central South University of Forestry Technology, 2012,32:1673-923x.
[13] 陆开宏,胡智勇,梁晶晶,等.富营养水体中2种水生植物的根际微生物群落特征 [J]. 中国环境科学, 2010,30(11):1508-1515.
[14] Xiang X M, Song C X, Li Y S, et al. Microorganism features of Typha latifolia and Phragmites australis at rhizosphere [J]. Journal of Environmental Protection Science, 2004,30(8):35-38.
[15] Marika T, Jaanis J, Jaak T, et al. Microbial biomass, activity and community composition in constructed wetlands [J]. Science of the Total Environment, 2009,407(13):3958-3971.
[16] Ren L J, Wu Y N, Ren N Q, et al. Microbial community structure in an integrated A/O reactor treating diluted livestock wastewater during start-up period [J]. Journal of Environmental Sciences, 22(5):656-662.
[17] 王 莹.污染河流中微生物群落结构的空间变化解析 [D]. 吉林: 东北师范大学, 2008.
[18] 殷 峻,闻 岳,周 琪.人工湿地中微生物生态的研究进展 [J].环境科学与技术, 2007,30(1):108-110.
[19] Rousseau D P L, Lesage E. Constructed wetlands for water reclamation [J]. Desalination, 2008,218(1):181-189.
[20] Greenway M. The role of constructed wetlands in secondary effluent treatment and water reuse in subtropical and arid Australia [J]. Ecological Engineering, 2005,25(5):501-509.
[21] 卜梦娇,冯雪冰,杨小静,等.北京市再生水补水公园湿地水生植物群落调查 [J]. 湿地科学, 2012,10(2):223-227.
[22] Crowe A U, Plant A L, Kermode A R. Effects of an industrial effluent on plant colonization and on the germination and post-germinative growth of seeds of terrestrial and aquatic plant species [J]. Environmental Pollution, 2002,117(1):179-189.
[23] Marsh T L. Terminal restriction fragment length polymorphism (T-RFLP): an emerging method for characterizing diversity among homologous populations of amplification products [J]. Current Opinion in Microbiology, 1999,2(3):323-327.
[24] 宋洪宁,杜秉海.环境因素对东平湖沉积物细菌群落结构的影响[J]. 微生物学报, 2010,50(8):1065-1071.
[25] Tipaynoa S, Kimb C G, Saa T. T-RFLP analysis of structural changes in soil bacterial communities in response to metal and metalloid contamination and initial phytoremediation [J]. Applied Soil Ecology, 2012,61:137–146.
[26] Ginige M P, Kekkonen A H, Morris C, et al. Bacterial community and groundwater quality changes in an anaerobic aquifer during groundwater recharge with aerobic recycled water [J]. FEMS Microbiology Ecology, 2013,85(3):553-567.
[27] 黄 艺,舒中亚.基于浮游细菌生物完整性指数的河流生态系统健康评价——以滇池流域为例 [J]. 环境科学, 2013,34(8): 3010-3018.
[28] Ding T, Palmer M W, Melcher U. Community terminal restriction fragment length polymorphisms reveal insights into the diversityand dynamics of leaf endophytic bacteria [J]. BMC Microbiology, 2013,13(1):1.
[29] Liu W T, Marsh T L, Cheng H, et al. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA [J]. Applied and Environmental Microbiology, 1997,63(11):4516-4522.
[30] Mayrand P E, Corcoran K P, Ziegle J S, et al. The use of fluorescence detection and internal lane standards to size PCR products automatically [J]. Applied and Theoretical Electrophoresis, 1992,3(1):1-11.
[31] Ziegle J S, Su Y, Corcoran K P, et al. Application of automated DNA sizing technology for genotyping microsatellite loci [J]. Genomics, 1992,14(4):1026-1031.
[32] Zhang R, Thiyagarajan V, Qian P Y. Evaluation of terminalrestriction fragment length polymorphism analysis in contrasting marine environments [J]. FEMS Microbiology Ecology, 2008,65(1):169–178.
[33] De Figueiredo D R, Pereira M J, Moura A, et al. Bacterial community composition over a dry winter in meso- and eutrophic Portuguese water bodies [J]. FEMS Microbiology Ecology, 2007, 59(3):638–650.
[34] Xiong J, Liu Y, Zhang H, et al. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau [J]. Environmental Microbiology, 2012,14(9):2457–2466.
[35] Shen C, Xiong J, Zhang H, et al. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain [J]. Soil Biology and Biochemistry, 2013,57:204-211.
[36] 任丽娟,何 聃,邢 鹏,等.湖泊水体细菌多样性及其生态功能研究进展 [J]. 生物多样性, 2013,21(4):421–432.
[37] Langenheder S, Lindström E S, Tranvik L J. Structure and function of bacterial communities emerging from different sources under identical conditions [J]. Applied and Environmental Microbiology, 2006,72:212–220.
[38] Yannarell A C, Triplett E W. Geographic and environmental sources of variation in lake bacterial community composition [J]. Applied and Environmental Microbiology, 2005,71:227–239.
[39] Yan Q M, Zhang X X, Zhang T, Fang H H P. Seasonal microbial community shift in a saline sewage treatment plant [J]. Frontiers of Environmental Science and Engineering in China, 2011,5(1): 40-47.
[40] Jiang H, Dong H, Yu B, et al. Microbial response to salinity change in Lake Chaka, a hypersaline lake on Tibetan plateau [J]. Environmental Microbiology, 2007,9(10):2603–2621.
[41] Herlemann D P R, Labrenz M, Jurgens K, et al. Transitions in bacterial communities along the 2000km salinity gradient of the Baltic Sea [J]. International Society for Microbial Ecology, 2011, 5:1571–1579.
[42] Hamdan L J, Jonas R B. Seasonal and interannual dynamics of free-living bacterioplankton and microbially labile organic carbon along the salinity gradient of the Potomac River [J]. Estuaries and Coasts, 2006,29(1):40-53.
[43] Wu Q L, Zwart G, Schauer M, et al. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan plateau [J]. Applied and Environmental Microbiology, 2006,72(8):5478-5485.
[44] Yan Q Y, Yu Y H, Feng W S. Plankton community composition in the Three Gorges Reservoir Region revealed by PCR-DGGE and its relationships with environmental factors [J]. Journal of Environmental Sciences, 2008,20(6):732-738.
[45] 孙 巍.东江微生物的群落结构及其在氨氮转化中的作用特点[D]. 广州:华南理工大学, 2011年.
[46] Fu T X, Wei K J, Xu G H. The research and application of bacillus in aquaculture [J]. Reservoir Fisheries, 2007,27(3):102-104.
[47] Gerner-Smidt P, Keiser-Nielsen H, Dorsch M, et al. Lautropia mirabilis gen. nov., sp. nov., a Gram-negative motile coccus with unusual morphology isolated from the human mouth [J]. Microbiology, 1994,140(7):1787-1797.
[48] Kisand V, Cuadros R, Wikner J. Phylogeny of culturable estuarine bacteria catabolizing riverine organic matter in the Northern Baltic Sea [J]. Applied and Environmental Microbiology, 2002,68(1):379-388.
[49] Nijburg J W, Laanbroek H J. The influence of Glyceria maxima and nitrate input on the composition and nitrate metabolism of the dissimilatory nitrate-reducing bacterial community [J]. FEMS Microbiology Ecology, 1997,22(1):57-63.
[50] Moore E R B, Tindall B J, Dos Santos V A P M, Pieper D H, Ramos J L, Palleroni N J. Nonmedical: pseudomonas [M]//The Prokaryotes. Springer New York, 2006:646-703.
[51] Andrianasolo E H, Goeger D, Gerwick W H. Mitsoamide: A cytotoxic linear lipopeptide from the Madagascar marine cyanobacterium Geitlerinema sp [J]. Pure and applied chemistry, 2007,79(4):593-602.
[52] Chen C, Ren N, Wang A, et al. Microbial community of granules in expanded granular sludge bed reactor for simultaneous biological removal of sulfate, nitrate and lactate [J]. Applied microbiology and biotechnology, 2008,79(6):1071-1077.
[53] Ubalde M C, Braña V, Sueiro F, et al. The versatility of Delftia sp. isolates as tools for bioremediation and biofertilization technologies [J]. Current microbiology, 2012,64(6):597-603.
[54] Eiler A, Bertilsson S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes [J]. Environmental Microbiology, 2004,6(12): 1228-1243.
[55] Figueiredo D R, Pereira M J, Moura A, et al. Bacterial community composition over a drywinter in meso- and eutrophic Portuguese water bodies [J]. FEMS Microbiology Ecology, 2007,59(3):638-650.
[56] 魏曼曼,陈新华,周洪波.深海热液喷口微生物群落研究进展 [J].海洋科学, 2012,36(6):113-121.
致谢:本实验的现场采样和实验工作由实验室同学赵霏、黄迪及马栋山等协助帮忙完成,在此表示感谢.
Influence of reclaimed water on bacterial community structure of cattail rhizosphere from riverine wetland.
HUANG Xing-ru1,2, ZHANG Qiong-qiong1,2, ZHANG Rui-jie1,2, GUO Xiao-yu1,2*(1.College of Resources Environment and Tourism, Capital Normal University, Beijing 100048, China;2.Urban Environmental Processes and Digital Modeling Laboratory, Beijing 100048, China). China Environmental Science, 2016,36(2):569~580
Abstract:Water reclamation and reuse have been actively promoted in Beijing, but the potential influences of reclaimed water on the microbial community structures are still poorly understood. Therefore, bacterial community structures in cattail rhizosphere between the samples of reclaimed water outfall and far from the reclaimed water outfall in the Mayu Wetland of Yongding River, Beijing were compared. Terminal restriction fragment length polymorphism (T-RFLP) was conducted to quantitatively detect the changes of bacterial community structures. Several statistical methods including one-way analysis of variance (ANOVA), spearman’s correlation analysis and canonical correspondence analysis (CCA) were united to find out which were the key environmental factors to drive the bacterial community structure shifts. The result showed that microbial richness, evenness and diversity decreased with the increase of the reclaimed water interference intensity. The diversity of Gammaproteobacteria, Deltaproteobacteria, Chloroflexi, Epsilonproteobacteria and Actinobacteria were decreased significantly near the reclaimed water outfall. Spearman’s correlation analysis indicated that pH, DO, TDS, ORP, Sal and NH4+-N play an important role in the diversity spatial variation of plant rhizosphere microbial community. CCA indicated that TN, TOC, and Cr、Ni、Cu were significantly correlated with microbial communities structures of the upstream of reclaimed water outfall. Plant rhizosphere bacterial communities near the outfall were significantly different due to the reclaimed water quality difference. Group IIa and IIb were mainly affected by pH and T、ORP、NH4+-N, respectively. While the bacterial communities in the downstream were also significantly correlated with TOC and some heavy metals due to water internal impurities and human activity influence.
Key words:reclaimed water;T-RFLP;bacterial community diversity;multivariate statistical analysis
作者简介:黄兴如(1988-),男,安徽阜阳人,首都师范大学硕士研究生,主要从事环境微生物研究.
基金项目:国家自然科学基金(40901281);北京市教育委员会科技计划面上项目(KM201310028012)
收稿日期:2015-07-25
中图分类号:X172
文献标识码:A
文章编号:1000-6923(2016)02-0569-12
