
大庆盐碱地九种植物根际土壤微生物群落结构及功能多样性
2016-04-13 00:59:58杜滢鑫谢宝明蔡洪生郭长虹
生态学报 2016年3期
关键词:盐碱地
杜滢鑫, 谢宝明, 蔡洪生, 唐 璐, 郭长虹
哈尔滨师范大学生命科学与技术学院, 分子细胞遗传与遗传育种黑龙江省重点实验室, 哈尔滨 150025
大庆盐碱地九种植物根际土壤微生物群落结构及功能多样性
杜滢鑫, 谢宝明, 蔡洪生, 唐璐, 郭长虹*
哈尔滨师范大学生命科学与技术学院, 分子细胞遗传与遗传育种黑龙江省重点实验室, 哈尔滨150025
摘要:盐碱土是陆地表面生态脆弱区域。它与荒漠化过程相伴而生,不但造成了资源的破坏、农业生产的巨大损失,而且还对生物圈和生态环境构成威胁。研究盐碱地植物根际土壤微生物群落的多样性,对于盐碱土壤的植被恢复和生态重建具有重要意义。运用PCR-DGGE技术和Biolog微平板法,对大庆盐碱地9种不同植物根际土壤微生物结构和功能的多样性进行了分析。结果表明,不同植物根际土壤微生物组成不同,同一科的植物具有相似的微生物组成。对11个克隆进行了序列测定,发现这一地区植物根际优势微生物菌群为变形菌门(Proteobacteria)和酸杆菌门(Acidobacteria)。利用Biolog微平板法分析了微生物群落功能多样性。结果表明,不同植物根际土壤细菌群落对底物碳源的代谢特征存在着一定的差异,其中豆科的野大豆根际土壤细菌对底物碳源的代谢能力最强。
关键词:盐碱地; 植物根际; 微生物群落; PCR-DGGE; Biolog微平板法
土地盐碱化是土地退化的主要形式,同时是生态环境的一种恶化现象,严重影响农业的发展。中国现有盐渍土地总面积为10000×104hm2[1]。大庆地区盐碱化总面积42.9955×104hm2,其中大庆盐碱化面积14.8687×104hm2,肇州盐碱化面积4.8113×104hm2,肇源盐碱化面积8.769×104hm2,林甸盐碱化面积4.6833×104hm2,杜蒙盐碱化面积9.8592×104hm2[2]。
盐生植物因为生境恶劣,生物量小,经济效益低,所以没有得到人们的足够重视,盐生植物的开发利用程度不够,严重影响了盐渍土的利用和改良。但是大多数盐生植物则是盐渍土上唯一能够生存的植物,它们对盐渍土的开发与利用,以及维持生态平衡起着至关重要的作用,它们的生态价值是不可以被忽略的。随着人口的增长和经济的发展以及改善生态环境的迫切需要,盐渍土资源和耐盐植物资源将会变得日益重要[3]。
土壤是许多土壤微生物的生存寄居场所,由于土壤有机质含量酸碱度水分及土质的不同,与此环境相适应的土壤微生物种类也千差万别,在同一地区,不同的植物根际土壤微生物种类和数量也不一样[4]。盐碱土壤中植物的根际微生物群落研究对于加强对盐生植物资源的开发和利用,以及改良盐碱土壤具有十分重要的意义。
本文从盐碱土壤中植物的根际微生物群落研究入手,以大庆盐碱地区不同植物根际土壤为实验材料,利用PCR-DGGE和Biolog微平板法分析,评价盐碱地不同植物根际微生物群落结构及功能多样性,为盐碱土壤的植被恢复奠定基础。
1研究区域
盐碱样地位于黑龙江省大庆市采油三厂第五矿区周围草地,地理为46°41′—46°42′N,124°59′—124°60′E。该地属于大陆性半干旱温带草原气候,降水量最大时期为7月份。土壤为盐碱土,pH值为8.7—9.3,主要优势植物有鹅观草(Roegneriakamoji)、羊草(Leymuschinensis)、虎尾草(Chlorisvirgata)、黄芪(Astragalusmembranaceus)、野大豆(Glycinesoja)、甘草(Glycyrrhizauralensis)、菊芋(Helianthustuberosus)、大籽蒿(Artemisiasieversiana)、碱地肤(Kochiascoparia)等。
2样品来源与研究方法
2.1样品来源
于2013年8月1日对盐碱样地进行土壤样品采集。在盐碱样地内随机选取3个样点,按五点采样法采集0—20 cm土壤样品,用铁锹将植物从土壤中连根挖出,将植物根际土壤自然抖落,混合均匀,用标本采集袋将植物装好,将土壤样品放在便携式冰盒中带回实验室,用于Biolog微平板法分析的土样取根际鲜土立即用于实验,用于DGGE分析的土样,放在-20 ℃冰箱保存。
2.2研究方法2.2.1土壤DNA的提取
提取方法在Zhou[5]的基础上进行了改进,取土壤样品0.25 g,置50 mL离心管中,偏磷酸钠(pH值8.5,含1% PVP)洗涤3次,搅拌均匀,10000 r/min离心5 min,转移沉淀至1.5 mL离心管中,贮存于-20 ℃冰箱。取沉淀,重悬于500 μL DNA提取缓冲液,加入溶菌酶(50 mg/mL)至1 mg/mL,颠倒混匀,37 ℃水浴1 h,加入蛋白酶K至100 μg/mL,65 ℃水浴1 h;中间反复冻融3次;加入等体积的氯仿/异戊醇抽提,12000 r/min离心10 min,取上清加入0.5倍体积PEG 8000,12000 r/min离心10 min,-20 ℃过夜,沉淀用70%乙醇洗涤,8000 r/min离心5 min,晾干,溶于100 μL pH值8.0 TE缓冲液。得到土壤总DNA。
2.2.2PCR扩增
PCR扩增的引物采用细菌通用性引物,序列是:B968FGC(5′ -CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGAACGCGAAGAACCTTAC-3′),B1401R(5′-GCGTGTGTACAAGACCC-3′)[6]。PCR的反应体系是50 uL,PCR反应条件为:94 ℃预变性5 min;94 ℃变性1 min,55 ℃退火1 min,72 ℃延伸1 min,30个循环;最后在72 ℃延伸7 min。扩增结果用2%的琼脂糖凝胶进行检测。
2.2.3PCR产物的DGGE
在Muyzer的方法进行优化[7]。DGGE采用BIO-RAD Dcode Universal Mutation Detection System进行,丙烯酰胺凝胶强度为8%,变性剂梯度为40%—60%,100%的变性剂含有7 mol/L尿素和40%的去离子甲酰胺。每孔加入10 μL的PCR产物和6 μL的6 × Loadding buffer,加样完成后,接通电泳电源,在70 V、60 ℃条件下电泳13 h。电泳结束后,DGGE凝胶放在SYBR Green中浸泡30 min,脱水10 min后,最后利用Bio-Rad凝胶成像系统进行拍照并分析。
2.2.4DGGE图谱分析
图谱分析采用Bio-Rad公司的凝胶定量软件Quantity One 4.6.5。对样品条带分析。根据DGGE图谱中条带分布用UPGMA(Unweighted pair group method Using arithmetic averages)进行聚类分析。
2.2.5条带的切胶、克隆和测序
切胶、克隆和测序主要参照王小芬[8]等的方法。扩增出来的产物送交到上海生工生物工程股份有限公司测序。将测序结果递交RDP(Ribosomal Database Project)数据库进行菌种鉴定。
2.2.6Biolog-ECO微平板分析
称取5 g土壤加入到装有45 mL灭菌NaCl溶液的三角瓶中(浓度为0.85%),然后在旋转振荡器上震荡30 min摇匀。土壤溶液依次稀释至10-3,向Biolog微平板(美国BIOLOG公司)上每孔加入150 μL土壤稀释液,然后在室温条件下(28 ℃),将微孔板放在保湿容器中避光培养,每隔24 h用酶标仪读取在590 nm(颜色+浊度)和750 nm(浊度)波长的数值。
单孔平均光密度(AWCD)计算按照Garland和Mills的方法[9],即AWCD(590—750)nm=∑(C590—750)/31,式中31为Biolog微平板上供试碳源的种类数。


图1 16S rDNA扩增片段的DGGE图谱Fig.1 DGGE profiles of the PCR products M:Marker,Ht:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
2.2.7数据分析
所有数据均用软件Excel和SPSS 19.0软件用Duncan检验方法进行数据分析处理和差异显著性分析(P< 0.05差异显著)。同列数据后具有相同字母者,表示在0.05水平上差异不显著。
3结果与分析
3.1PCR-DGGE结果分析3.1.1盐碱地植物根际土壤细菌PCR-DGGE分析
对大庆盐碱地区9种不同植物根际土壤细菌多样性进行了DGGE分析(图1)。结果表明,DGGE条带数目、强度、均匀度都存在不同的差异。大庆盐碱地区九种不同植物根际土壤样品之间具有公共的条带,说明供试土壤根际微生物之间可能存在一些共有的细菌类群,然而有些公共条带的亮度存在差异,表明不同植物根际细菌具有数量上的差异。
从图1和表1可以看出,1、2、4、8、10、11是研究样本的共有细菌,其中3菊芋、大籽蒿、甘草和黄芪样本中不存在,其他样本中均存在;5甘草和羊草样本中不存在,其他样本中均存在;6碱地肤中不存在,其他样本中均存在;7菊芋、大籽蒿和碱地肤中不存在,其他样本中都存在。
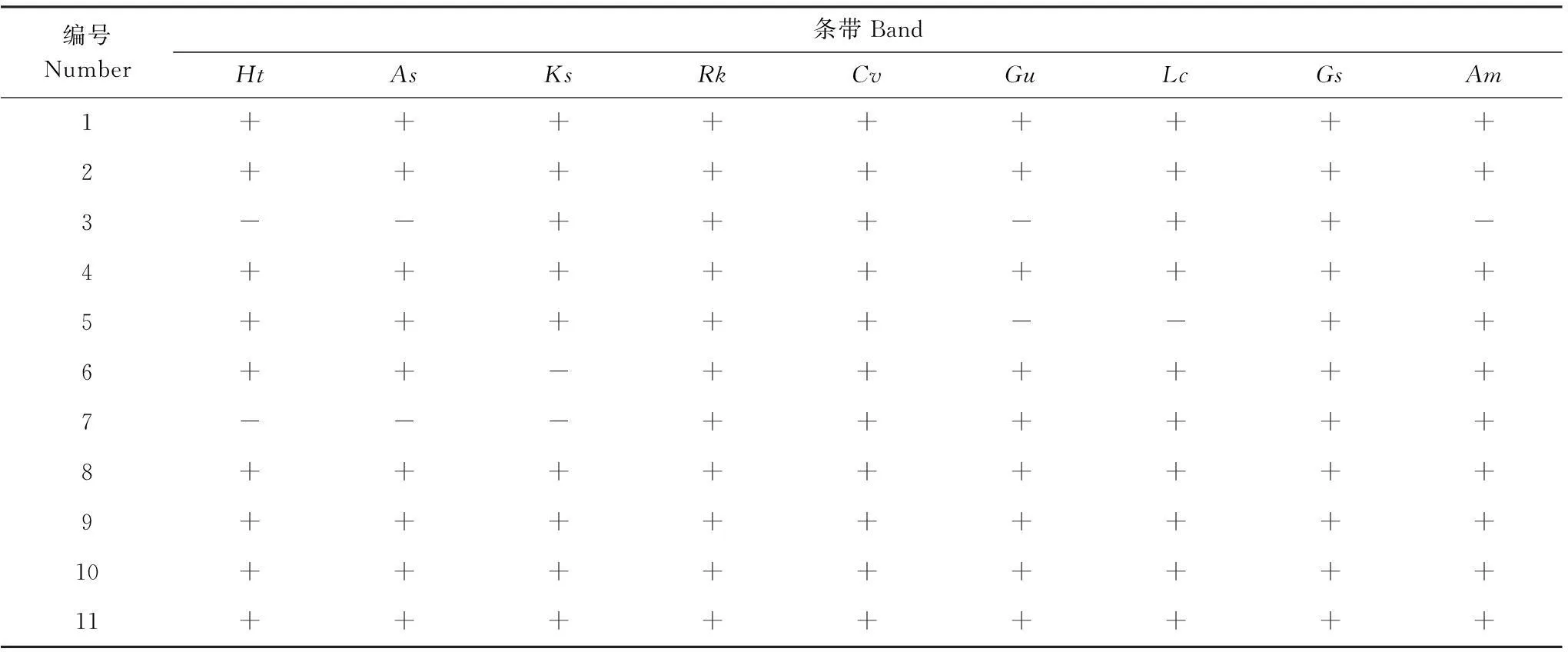
表1 土壤细菌PCR-DGGE电泳图谱条带分析
Ht:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
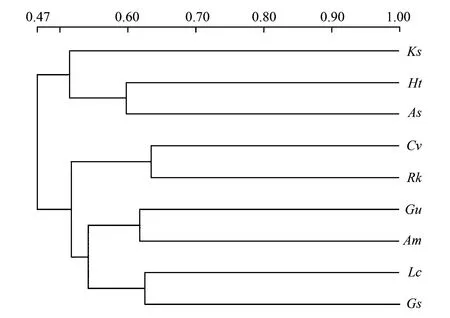
图2 DGGE图谱聚类分析Fig.2 Cluster analysis of DGGE banding patterns Ht:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
3.1.2DGGE聚类分析
运用Bio-Rad公司Quantity One分析软件绘制出不同植物根际微生物之间的遗传簇关系(图2)和遗传距离与群落差异(表2)。根据群体间不同的遗传距离,对其进行聚类分析(UPGMA),结果表明,当取值水平在0.55以上时,菊芋和大籽蒿,虎尾草与鹅观草,甘草与黄芪根际微生物分别属于同一簇。说明同一科植物根际微生物的类群比较接近。
3.1.3DGGE条带测序分析
将测序结果递交RDP数据库进行鉴定,结果表明,1、2、4为酸杆菌门(Acidobacteria),5、8、9、10、11为变形菌门(Proteobacteria),其他为未知菌种。
3.2Biolog微平板结果分析
3.2.1不同植物根际土壤微生物利用总碳源的动力学特征

表2 不同植物的遗传距离和细菌群落差异
Ht:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
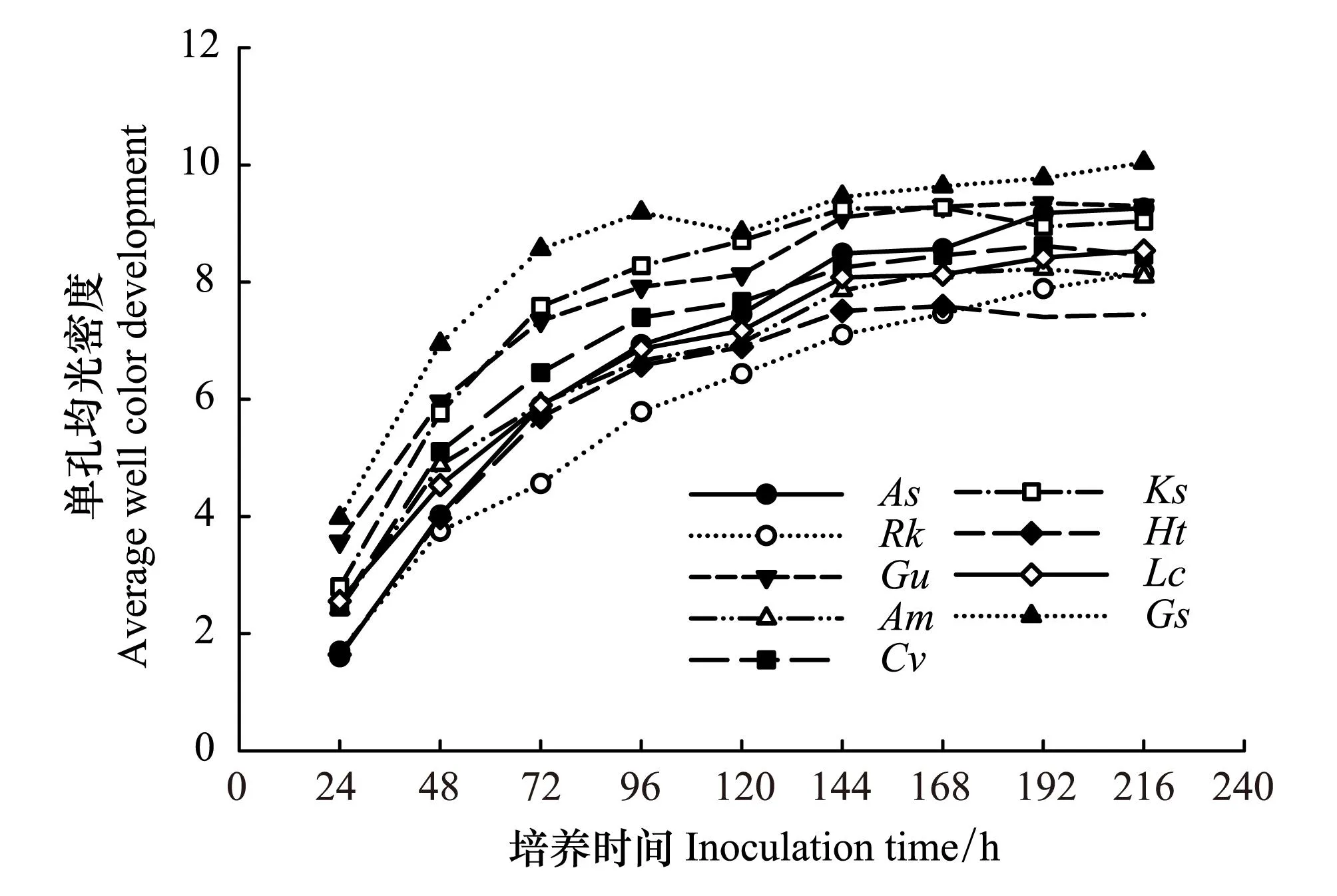
图3 不同植物根际土壤微生物AWCD随时间的变化曲线 Fig.3 Ariation in AWCD of rhizosphere soil microbial communities over time for different plantsHt:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
对9种植物根际土壤微生物进行了Biolog微平板分析(图3)。AWCD曲线反映土壤细菌在Biolog微平板中的生长情况,通常认为,曲线变化幅度越大的样品碳源利用能力较高,也具有比较高的丰富度。结果表明,9种植物中根际微生物对碳源利用能力最强的是野大豆,其他植物对碳源的利用能力比较接近。
3.2.2土壤微生物功能多样性的主成分分析
进一步对不同植物土壤细菌群落底物碳源代谢主成分进行了分析(图4)。结果表明,野大豆和碱地肤与其他植物没有交集,说明野大豆和碱地肤根际土壤细菌群落对底物碳源的代谢特征与其他植物有显著差异。鹅观草除了与菊芋有交集外,同其他植物没有交集,说明鹅观草根际土壤细菌群落对底物碳源的代谢特征只与菊芋无明显差异,同其他植物都有明显差异。
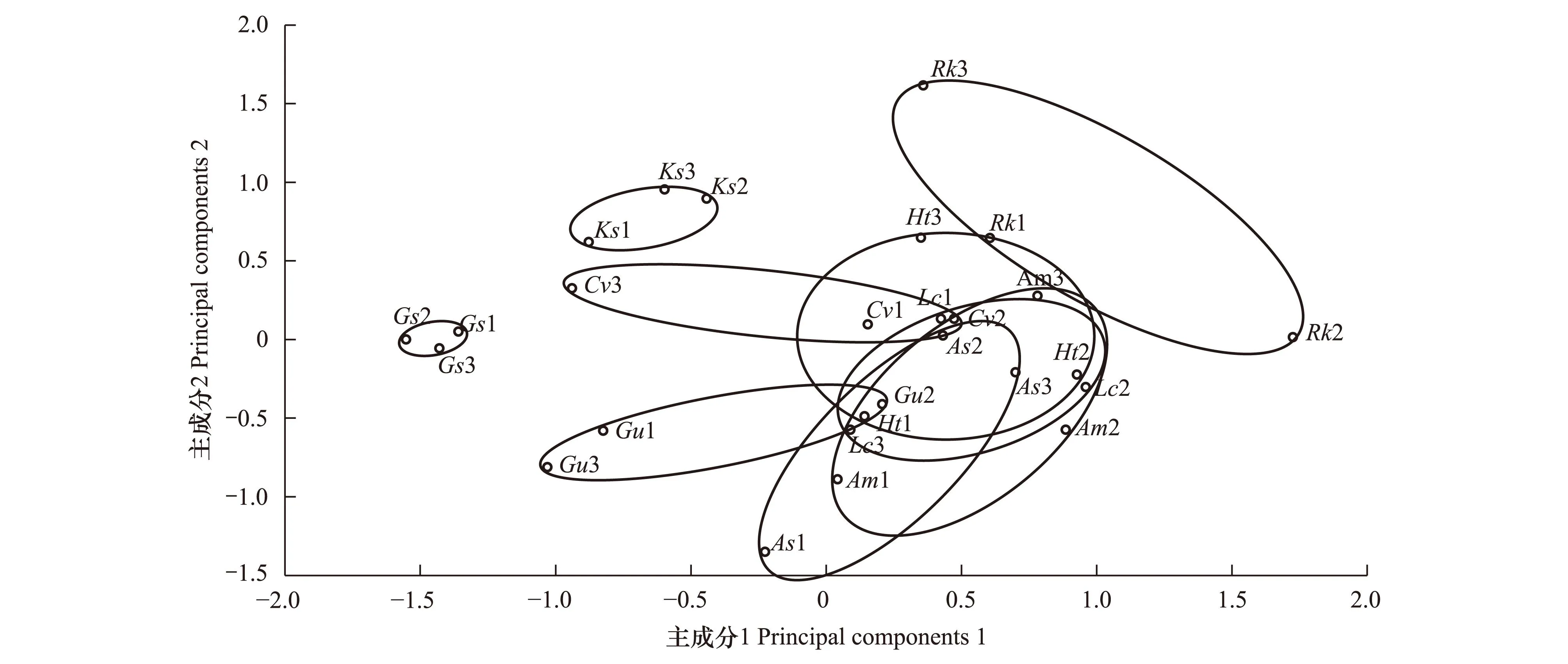
图4 不同植物根际土壤细菌群落的主成分分析Fig.4 Principal component analysis (PCA) of soil bacteria communities in rhizosphere for different plantsHt1-3:菊芋,As1-3:大籽蒿,Ks1-3:碱地肤,Rk1-3:鹅观草,Cv1-3:虎尾草,Gu1-3:甘草,Lc1-3:羊草,Gs1-3:野大豆,Am1-3:黄芪
3.2.3土壤微生物群落多样性指数分析
对不同植物根际土壤微生物在培养96 h的多样性指数进行了分析(表3)。Shannon多样性指数是研究个体数及群落种群数和分布均匀度的综合指标,它是目前应用最广泛的群落多样性指数之一。Simpson指数较多反映了微生物群落中最常见的物种优势度,MicIntosh指数则是群落物种均一性的度量。从表3可以看出,Shannon多样性指数与Simpson指数均以野大豆土壤样本最高,鹅观草土壤样本最低,不同植物根际土壤样本间存在一定的差异。MicIntosh指数的变化与Shannon多样性指数正相反,鹅观草土壤样本最高,野大豆土壤样本最低。

表3 不同植物根际土壤微生物群落多样性指数
不同字母表示统计学上的差异性(P< 0.05);Ht:菊芋,As:大籽蒿,Ks:碱地肤,Rk:鹅观草,Cv:虎尾草,Gu:甘草,Lc:羊草,Gs:野大豆,Am:黄芪
4讨论
土壤中绝大多数的微生物是不可培养的[11]。所以,传统的稀释平板涂布方法不能真实和准确的反应出土壤中微生物的结构组成,这就需要运用更先进的分子生物学方法来了解土壤中微生物的结构和组成[12]。
本文通过DGGE方法,研究了大庆盐碱地植物根际土壤群落结构多样性,结果表明,不同的植物之间土壤微生物群落结构相似性比较高,但也存在着一些差异。DGGE指纹图谱的聚类分析显示,同一科的植物,它们的根际土壤微生物群落结构归为同一簇,表明植物本身对根际土壤微生物群落有一定的调节作用。DGGE图谱中的优势条带进行测序结果表明,其中共有的1、2、4细菌属于酸杆菌门(Acidobacteria),5、8、9、10、11细菌属于变形菌门(Proteobacteria),具有差异的3、6、7细菌均为未知菌种。这与Fierer等[13]和Deangelis等[14]的研究类似,他们推测变形菌门在各种植物根际微生物占优势菌群的原因是它们在各种植物的根际中增长的速度很快。同时,本研究出现了酸杆菌门的菌群与Kielak等[15]和Yergeau等[16]的研究相类似。他们认为酸杆菌门细菌是微生物生态系统中稳定的组成部分,因为它们所需的营养物质很少或者营养物质不变。Duineveld等[17]也曾证明这些是由它们贫营养的性质所决定的,这使得它们能慢慢的应对植物根环境的变化,包括那些由根分泌物产生的影响。这与郑贺云等[18]的研究有所不同,郑贺云等发现29个序列属于不可培养的微生物,43个序列分属粘球菌目(Myxococcales)、假单胞菌目(Pseudomonadales)、根瘤菌目(Rhizobiales)、芽孢杆菌目(Bacillales)、伯克氏菌目(Burkholderiales)、放线菌目(Actinomycetales)、海洋螺菌目(Oceanospirillales)、黄杆菌目(Flavobacteriales)、交替单胞菌目(Alteromonadales),21个属,因为研究的地域不同,所以出现不同的研究结果。
Biolog微平板法广泛用于评价土壤微生物群落的功能多样性。微生物功能多样性对同一环境下不同植物根际微生物群落具有重要意义。通过Biolog微平板AWCD分析,可以看出豆科植物根际土壤微生物群落对碳底物利用的能力明显强于其他科植物,滕应等[19]在研究复垦红壤中牧草根际微生物群落时,推测植物向根际土壤分泌的碳水化合物越多,根际微生物对碳底物利用的能力越强。本研究的结果表明,豆科植物野大豆向根际土壤分泌的碳水化合物要超过其他植物,同一科的植物根际土壤细菌群落对底物碳源的代谢特征比较相似(如黄芪和甘草,虎尾草和羊草,菊芋和大籽蒿),不同科植物根际土壤细菌群落对底物碳源的代谢特征存在着一定的差异(如碱地肤与其他植物)。Shannon多样性指数反应物种的丰富度,Simpson指数反应常见物种的优势度,野大豆无论在物种的丰富度,还是物种的优势度上都处于最高数值。豆科植物黄芪和甘草的Shannon和Simpson指数也相对较高,这与Perez-Montan等[20]的研究结果相似,他认为豆科植物和根瘤菌共生系统可以很好的完成生物固氮,从而提高根际微生物的数量。
DGGE技术对根际土壤进行检测时存在一些难题,微生物群落中数量小于1%的种群不能采用DGGE技术进行检测分析[7]。两个微生物16S rRNA的GC含量相同,可是序列组成却不同,如果从DGGE图谱上看,它们都是同一个条带,这就不能准确的分析微生物群落的多样性,容易产生错误的分析结果[21]。所以,为了降低由于分析方法产生的误差,分析微生物群落多样性时可以将几种不同的分析方法结合使用[22]。目前许多研究正在利用宏基因组学的方法分析微生物的多样性,它是直接提取全部微生物的DNA,构建宏基因组文库,利用基因组学研究微生物的群落功能和遗传组成[23],是一种研究微生物多样性的新理念和新方法,在未来可能会有广泛的应用。
本研究运用PCR-DGGE技术和Biolog微平板法,分析了大庆盐碱地区9种不同植物根际微生物结构和功能的多样性,为盐碱地土壤的开发利用和生物修复提供了重要参考。
参考文献(References):
[1]张宝贤, 王光明, 谭德云, 刘红光, 杨云峰, 孙丽娟. 蓖麻杂交种耐盐碱能力评价试验初报. 中国农学通报, 2012, 28(33): 88- 92.
[2]陈建军, 张树文, 陈静, 唐俊梅. 大庆市土地盐碱化遥感监测与动态分析. 干旱区资源与环境, 2003, 17(4): 101- 106.
[3]刘阳春, 何文寿, 何进智, 沈振荣. 盐碱地改良利用研究进展. 农业科学研究, 2007, 28(2): 68- 71.
[4]李凤霞, 郭永忠, 许兴. 盐碱地土壤微生物生态特征研究进展. 安徽农业科学, 2011, 39(23): 14065- 14067, 14174- 14174.
[5]Zhou J, Bruns M A, Tiedje J M. DNA recovery from soils of diverse composition. Applied and Environmental Microbiology, 1996, 62(2): 316- 322.
[6]Heuer H, Smalla K. Evaluation of community-level catabolic profiling using BIOLOG GN microplates to study microbial community changes in potato phyllosphere. Journal of Microbiological Methods, 1997, 30(1): 49- 61.
[7]Muyzer G, De Waal E C, Uitterlinden A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Applied and Environmental Microbiology, 1993, 59(3): 695- 700.
[8]王小芬, 王伟东, 高丽娟, 崔宗均. 变性梯度凝胶电泳在环境微生物研究中的应用详解. 中国农业大学学报, 2006, 11(5): 1- 7.
[9]Garland J L, Mills A L. Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Applied and Environmental Microbiology, 1991, 57(8): 2351- 2359.
[10]杨永华, 姚健, 华晓梅. 农药污染对土壤微生物群落功能多样性的影响. 微生物学杂志, 2000, 20(2): 23- 25.
[11]Amann R I, Ludwig W, Schleifer K H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews, 1995, 59(1): 143- 169.
[12]Pace N R. A molecular view of microbial diversity and the biosphere. Science, 1997, 276(5313): 734- 740.
[13]Fierer N, Bradford M A, Jackson R B. Toward an ecological classification of soil bacteria. Ecology, 2007, 88(6): 1354- 1364.
[14]Deangelis K M, Brodie E L, Desantis T Z. Selective progressive response of soil microbial community to wild oat roots. International Society for Microbial Ecology Journal, 2009, 3(2): 168- 178.
[15]Kielak A, Pijl A S, Veen J A, Kowalchuk G A. Differences in vegetation composition and plant species identity lead to only minor changes in soil-borne microbial communities in a former arable field. FFFS Microbiology Ecology, 2008, 63(3): 372- 382.
[16]Yergeau E, Schoondermark-Stolk S A, Brodie E L, Déjean S, DeSantis T Z, Gonçalves O, Piceno Y M, Andersen G L, Kowalchuk G A. Environmental microarray analyses of Antarctic soil microbial communities. International Society for Microbial Ecology Journal, 2009, 3(3): 340- 351.
[17]Duineveld B M, Rosado A S, van Elsas J D, van Veen J A. Analysis of the dynamics of bacterial communities in the rhizosphere of the chrysanthemum via denaturing gradient gel electrophoresis and substrate utilization patterns. Applied and Environmental Microbiology, 1998, 64(12): 4950- 4957.
[18]郑贺云, 黎志坤, 李超, 张鲜姣, 胡建伟, 朱红惠. 新疆阿克苏地区盐碱地细菌类群多样性及优势菌群分析. 微生物学通报, 2012, 39(7): 1031- 1043.
[19]滕应, 黄昌勇, 龙健, 姚槐应. 复垦红壤中牧草根际微生物群落功能多样性. 中国环境科学, 2003, 23(3): 295- 299.
[21]周小奇, 王艳芬, 蔡莹, 黄祥忠, 郝彦宾, 田建卿, 柴团耀. 内蒙古典型草原细菌群落结构的PCR-DGGE检测. 生态学报, 2007, 27(5): 1674- 1689.
[22]吴永胜, 马万里, 李浩, 卢萍, 吕桂芬. 内蒙古退化荒漠草原土壤细菌群落结构特征. 生态学报, 2010, 30(23): 6355- 6362.
[23]李慧, 何晶晶, 张颖, 徐慧, 陈冠雄. 宏基因组技术在开发未培养环境微生物基因资源中的应用. 生态学报, 2008, 28(4): 1762- 1773.
Structural and functional diversity of rhizosphere microbial community of nine plant species in the Daqing Saline-alkali soil region
DU Yingxin, XIE Baoming, CAI Hongsheng, TANG Lu, GUO Changhong*
LaboratoryofMolecularCytogeneticsandGeneticBreedingofHeilongjiangProvince,CollegeofLifeScienceandTechnology,HarbinNormalUniversity,Heilongjiang150025,China
Abstract:Saline-alkali soils are particularly ecologically fragile. They generally form following desertification, which destroys natural resources and causes heavy losses to agriculture production. Daqing, a city in the northern province of Heilongjiang, is renowned for its vast oil reserves. Exploitation of these reserves has led to soil contamination by petroleum-derived products, and this is becoming serious ecological problem in this city. Saline-alkali soil is another factor that contributes to rendering most of its agricultural land unusable. In recent years, bioremediation, especially microbial remediation, has been a global focus of research. It is more effective, safer, and more environmentally friendly than other remediation strategies, such as chemical or physical methods. The rhizosphere—the layer of soil influenced by plant roots—is much richer in microbial diversity than the surrounding bulk soil. Exploring the diversity of plant rhizosphere microbial communities in Saline-alkali soils can therefore provide a scientific basis for vegetation restoration and ecological reconstruction in this region. This study focused on plant rhizosphere microbial communities in Saline-alkali soils in the Daqing region. Using Biolog EcoPlate methods, denaturing gradient gel electrophoresis (DGGE) analysis, and subsequent DNA sequencing, we investigated the structural and functional diversity of rhizosphere microbial communities associated with nine plant species in Daqing Saline-alkali soils. The structure of the rhizosphere microbial community associated with each plant species was analyzed by DGGE. Plants in the same family tended to have similar rhizosphere microbial community compositions. Rhizosphere bacteria were dominated by Proteobacteria andAcidobacteria,based on the analysis of 16S rRNA. Pairwise rhizosphere population genetic distances between plant species were calculated using Quantity One software (Bio-Rad Laboratories). In combination with clustering analysis based on the unweighted pair group method with arithmetic mean, it was confirmed that rhizosphere microbial communities reflect relationships among the plant species tested. Biolog EcoPlate was used to investigate the functional diversity of rhizosphere microbial communities. This method is more sensitive to changes in the environment than the other methods such as phospholipid fatty-acid analysis. Changes in functional diversity patterns can be statistically analyzed via principle component analysis of average well color development data. The capacity of rhizobacteria to metabolize carbon sources was higher in communities associated with wild soybean than in other plant species tested. The data also suggest that rhizobacteria from different plant species have distinct carbon metabolism characteristics. While there are limitations in the methods used in this study, they nevertheless provided useful information. Metagenomics—the study of genetic material recovered directly from environmental samples—is becoming increasingly popular as a research tool. It provides a powerful lens for viewing the microbial world that has the potential to revolutionize the understanding of the entire living world. The results of this study offer advanced insights in the field of microbial ecology and provide a theoretical basis for future practical applications.
Key Words:saline-alkali soil; rhizosphere; microbial community; PCR-DGGE; biolog-ECO
DOI:10.5846/stxb201404020621
*通讯作者
Corresponding author.E-mail: kaku3008@126.com
收稿日期:2014- 04- 02; 网络出版日期:2015- 06- 12
基金项目:国家自然科学基金(31170479, 31470571); 国家科技支撑计划项目(2011BAD17B04-2-1); 黑龙江省科技攻关计划项目(GC12B304)
杜滢鑫, 谢宝明, 蔡洪生, 唐璐, 郭长虹.大庆盐碱地九种植物根际土壤微生物群落结构及功能多样性.生态学报,2016,36(3):740- 747.
Du Y X, Xie B M, Cai H S, Tang Lu, Guo C H.Structural and functional diversity of rhizosphere microbial community of nine plant species in the Daqing Saline-alkali soil region.Acta Ecologica Sinica,2016,36(3):740- 747.
猜你喜欢
军事文摘(2024年6期)2024-02-29 09:59:12
河北画报(2020年21期)2020-12-14 03:16:10
河北果树(2020年1期)2020-02-09 12:31:34
河北果树(2020年1期)2020-01-09 06:59:50
新疆农垦科技(2014年10期)2014-02-28 19:21:10
中国青年(1966年2期)1966-08-17 03:19:34
