
NADPH oxidase 2 does not contribute to early reperfusion-associated reactive oxygen species generation following transient focal cerebral ischemia
2016-02-09 05:17YuanZhangTingWangKeYangJiXuJianmingWuWenlanLiu1CentralLaboratoryShenzhenSecondPeopleHospitalFirstAffiliatedHospitalofShenzhenUniversityShenzhenGuangdongProvinceChinaShenzhenKeyLaboratoryofNeurosurgeryShenzhenSecondPeop
中国神经再生研究(英文版) 2016年11期
Yuan Zhang, Ting Wang, Ke Yang, Ji Xu Jian-ming Wu, Wen-lan Liu1 Central Laboratory, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, Guangdong Province, China Shenzhen Key Laboratory of Neurosurgery, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, Guangdong Province, China Department of Pathophysiology, Baotou Medical College, Baotou, Inner Mongolia Autonomous Region, China Graduate School of Guangzhou Medical University, Guangzhou, Guangdong Province, China Department of Neurosurgery, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, GuangdongProvince, China
NADPH oxidase 2 does not contribute to early reperfusion-associated reactive oxygen species generation following transient focal cerebral ischemia
Yuan Zhang1,2,3, Ting Wang1,2,4, Ke Yang1,2,4, Ji Xu1,2, Jian-ming Wu5, Wen-lan Liu1,2,*
1 Central Laboratory, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, Guangdong Province, China
2 Shenzhen Key Laboratory of Neurosurgery, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, Guangdong Province, China
3 Department of Pathophysiology, Baotou Medical College, Baotou, Inner Mongolia Autonomous Region, China
4 Graduate School of Guangzhou Medical University, Guangzhou, Guangdong Province, China
5 Department of Neurosurgery, Shenzhen Second People’s Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, Guangdong
Province, China
How to cite this article:Zhang Y, Wang T, Yang K, Xu J, Wu JM, Liu WL (2016) NADPH oxidase 2 does not contribute to early reperfusion-associated reactive oxygen species generation following transient focal cerebral ischemia. Neural Regen Res 11(11):1773-1778.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:The work was financially supported by grants from Shenzhen Science and Technology Innovation Commission of China, No. JCYJ20150330102401097, KQCX20140521101427034, JCYJ20140414170821291, and China Postdoctoral Science Foundation, No. 2015M572388.
Graphical Abstract
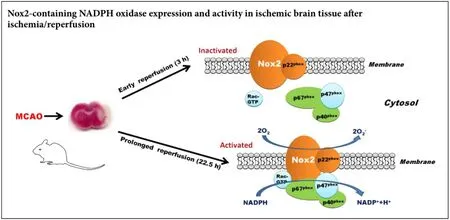
Excess production of reactive oxygen species (ROS) critically contributes to occurrence of reperfusion injury, the paradoxical response of ischemic brain tissue to restoration of cerebral blood flow. However, the enzymatic sources of ROS generation remain to be unclear. This study examined Nox2-containing NADPH oxidase (Nox2) expression and its activity in ischemic brain tissue following post-ischemic reperfusion to clarify the mechanism of enzymatic reaction of ROS. Male Sprague-Dawley rats were subjected to 90-minute middle cerebral artery occlusion, followed by 3 or 22.5 hours of reperfusion. Quantitative reverse transcriptase PCR and western blot assay were performed to measure mRNA and protein expression of Nox2. Lucigenin fluorescence assays were performed to assess Nox activity. Our data showed that Nox2 mRNA and protein expression levels were significantly increased (3.7-fold for mRNA and 3.6-fold for protein) in ischemic brain tissue at 22.5 hours but not at 3 hours following post-ischemic reperfusion. Similar results were obtained for the changes of NADPH oxidase activity in ischemic cerebral tissue at the two reperfusion time points. Our results suggest that Nox2 may not contribute to the early burst of reperfusion-related ROS generation, but is rather an important source of ROS generation during prolonged reperfusion.
nerve regeneration; NADPH oxidase; cerebral ischemia; Nox family; reactive oxygen species; reperfusion; central nervous system; stroke; blood flow; neural regeneration
Introduction
Timely restoration of cerebral blood flow (reperfusion) is the most effective treatment strategy for acute ischemic stroke. However, reperfusion can also exacerbate ischemic brain injury through a mechanism called “reperfusion injury”, which results in a paradoxical enlargement of infarct size and worse outcome when compared to the damage caused by permanent cerebral ischemia (without reperfusion) (Jung et al., 2010). To maximize the beneficial influence of reperfusion on tissue recovery, great efforts have been made to define the molecular and cellular basis of this unique injury response following postischemic reperfusion. Excessive production of reactive oxygen species (ROS) derived from the imbalance between the rate of ROS generation and tissue’s ability to detoxify these reactive species has been widely accepted as a key mediator for reperfusion injury in ischemic stroke (Amaro and Chamorro, 2011; Tang et al., 2014), however, the sources of reperfusion-associated ROS generation remain to be defined.
The Nox family of NADPH oxidases are multi-subunit enzymes including membrane bound subunits, cytoplasmic subunits, and G protein, which catalyze the reduction of molecular oxygen and oxidation of NADPH to generate superoxide anion (Babior, 2004). Among the five Nox family proteins (Nox1–5), Nox2 (formerly known as gp91phox) is originally found in phagocytic cells. Besides its expression in phagocytes, Nox2 is also extensively expressed in the central nervous system (CNS) and contributes to ROS generation in a variety of CNS pathological conditions including ischemic stroke (Serrano et al., 2003; Abramov et al., 2005; Manea et al., 2015; Rodriguez-Perez et al., 2015). In models of stroke, Nox2 and several other subunits have been found to be increased in ischemic brain tissue during postischemic reperfusion (Beske and Jackson, 2012; Kleikers et al., 2012; Kahles and Brandes, 2013), and inhibition of Nox2 with pharmacological inhibitors or genetic approaches (knock-out or knock-down) results in reduced ROS generation and smaller infarct size (Chang et al., 2011; Knox et al., 2014). These studies clearly support an important role of Nox2 in reperfusion-associated injury to the ischemic brain. However, the temporal profile of Nox2 expression is not fully characterized in the brain during postichemic reperfusion.
Restoration of blood flow to the ischemic brain triggers an early burst of ROS generation and sustained ROS generation during prolonged reperfusion, which critically contributes to reperfusion associated ischemic neuronal damage (Thompson et al., 2012; Rodrigo et al., 2013). Although several mechanisms, such as mitochondrial dysfunction (Sanderson et al., 2013), xanthine oxidase activation (Ono et al., 2009), neutrophil infiltration (Liou et al., 2003), and Rac1 GTPase induction (Ozaki et al., 2000) have been proposed to mediate reperfusion-associated ROS generation within ischemic brain tissue, the enzymatic sources of ROS generation at different stages of reperfusion have not yet been characterized.
In this study, we investigated the implication of Nox2 in reperfusion-induced ROS generation during the early and late phases of reperfusion in rats after transient focal cerebral ischemia. The data demonstrated that Nox2 was upregulated in ischemic brain tissue at 22.5 hours but not at 3 hours of reperfusion following 90-minute middle cerebral artery occlusion (MCAO), implicating a role for Nox2 in late reperfusion- but not early reperfusion-associated injury in ischemic stroke.
Materials and Methods
Animals
Forty-eight male Sprague-Dawley rats (SCXK (Yue) 2011-0015, Laboratory Animal Center of Southern Medical University, China) weighing 280–320 g were anesthetized with 4% isoflurane for surgical induction and their body temperature was maintained at 37.5 ± 0.5°C during the surgical procedure. All animal procedures were approved by the Laboratory Animal Care and Use Committee of Shenzhen University, China.
Middle cerebral artery occlusion/reperfusion (MCAO/R)According to a previous report (Liu et al., 2008), MCAO was induced by proximal occlusion of the right middle cerebral artery with a 4.0 monofilament nylon suture with a silicon coated tip. After 90-minute occlusion, the monofilament was carefully removed to establish reperfusion. Then, the rats were returned to their cages for 3 hours or 22.5 hours before sacrifice. Forty-eight MCAO/R rats were randomly divided into two groups: 90 min-I + 22.5 h-R group (90-minuate ischemia followed by 22.5-hour reperfusion,n= 24) and 90 min-I + 3 h-R group (90-minute ischemia followed by 3-hour reperfusion,n= 24). Eight MCAO/R rats were respectively used for each assay. For all rats used in this study, successful MCAO was confirmed by 2,3,5-triphenyltetrazolium chloride (TTC) staining as we described previously (Liu et al., 2012), and pale white region was considered as the area of tissue infarction in the ischemic hemispheres.
Tissue preparation
Emerging evidence suggests that blood-brain barrier damage occurred within the 3.0–4.5 hours thrombolytic time window (Hacke et al., 2008), so the rats were exposed to early-phase (3-hour) reperfusion and late-phase (22.5-hour) reperfusion respectively after 90-minute ischemia to assay Nox2 expression pattern in early reperfusion- and late reperfusion-associated injury in ischemic stroke. After 3-hour (early phase) or 22.5-hour (late phase) reperfusion, rats were sacrificed and brain tissues were collected and placed in ice-cold PBS. An 8-mm-thick brain region 3 mm away from the tip of the frontal lobe were then cut into eight 1-mm-thick coronary slices, which contained the main infarction area, indicative of success in MCAO as confirmed by 2% TTC staining, according to our previous study (Liu et al., 2004). After removal of meninges, non-ischemic and ischemic tissues were collected for detecting Nox2 protein and mRNA expression as well as NADPH oxidase activity.

Figure 1 Expression of Nox2 protein in ischemic brain tissue after 90-minute middle cerebral artery occlusion followed by 3 or 22.5 hours of reperfusion.

Figure 2 Nox2 mRNA expression in the ischemic brain tissue after 90-minute middle cerebral artery occlusion followed by 3 or 22.5 hours of reperfusion.
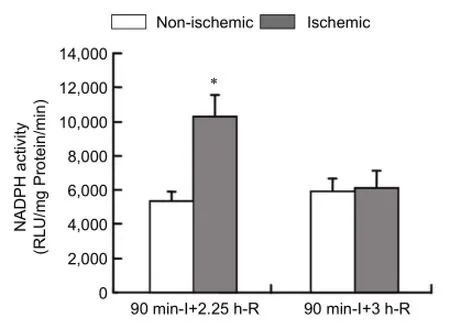
Figure 3 NADPH activity in ischemic brain tissue after 90-minute middle cerebral artery occlusion followed by 3 or 22.5 hours of reperfusion.
Quantitative reverse transcription-polymerase chain reaction
Non-ischemic and ischemic brain tissues were collected after 3-hour or 22.5-hour reperfusion, and total RNA was isolated from brain tissue and homogenized using Trizol reagents (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s protocols. RNA samples (2 μg) were reverse-transcribed to generate first-strand cDNA using TaqMan® Reverse Transcription Kits (Thermo Fisher Scientific, Waltham, MA). Reverse-transcribed products (0.5 μL) were amplified with the Vii7 real-time PCR System (Applied Biosystems, Foster City, CA, USA) in a 10 μL final reaction volume using SYBR® Green PCR Master Mix (Applied Biosystems) and the condition comprised an initial incubation at 95°C for 10 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing and extension at 60°C for 1 minute. Primer sequences and the length of amplified product were as follows: rat Nox2 forward: 5′-CAG CCT GCC TGA ATT TCA ACT-3′ and reverse: 5′-GGA GAG GAG ATT CCG ACA CAC T-3′, 62 bp; rat rpl 32 forward: 5′- GTG AAG CCC AAG ATC GTC-3′ and reverse: 5′-GAA CAC AAA ACA GGC ACA C-3′, 412 bp. The fluorescence threshold value (Ct value) was calculated using the ABI Vii7 Real-time PCR system software (Applied Biosystems, Foster City, CA, USA). Ct values were normalized to rpl 32 and the relative mRNA expression levels were determined by the 2−ΔΔCtquantification method, where ΔΔCt =ΔCt(target sample) −ΔCt(reference sample).
Western blot analysis for Nox2 protein expression
Non-ischemic and ischemic brain tissues were collected and then homogenized in RIPA lysis buffer (50 mM Tris-base, 150 mM NaCl, 2 mM EDTA, 10% glycerol, 1% TritonX-100, 1 mM phenylmethylsulfonyl fluoride) containing 1 mM Na3VO4and protease inhibitor cocktail (Sigma-Aldrich) at 4°C. The tissue lysates were centrifugedfor 15 minutes at 12,000 ×gat 4°C and protein concentrations were determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The cell lysates were subjected to electrophoresis on 10–12% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene difluoride membrane. The membranes were blocked in Tris-buffered saline with 0.1% Tween-20 containing 5% nonfat milk and membranes were then incubated overnight with monoclonal anti-Nox2 antibody (1:1,000; BD Transduction Laboratories, Forest Grove, OR, USA), The membranes were washed and incubated for 1 hour with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:1,000, Jackson ImmunoResearch, West Grove, PA, USA). The protein was detected using a chemiluminescence kit (Thermo Fisher Scientific) according to the manufacturer’s instructions, and the bands were visualized and quantified on a chemiluminescence and fluorescence gel Imaging System (Alliance, Uvitec Cambridge, UK). Nox2 protein expression was standardized to an equivalent β-actin protein amount and expressed as a ratio of band intensity (gray value) of ischemic brain tissue to non-ischemic brain tissue.
NADPH oxidase activity assay
NADPH oxidase activity was analyzed by the lucigenin-enhanced chemiluminescence method as described previously (Ostrowski et al., 2006). In brief, ischemic brain tissue was collected and then homogenized in ice-cold modified Krebs HEPES buffer containing NaCl 119 mM, HEPES 20 mM, KCl 4.6 mM, MgSO41.0 mM, CaCl21.2 mM, Na2HPO40.15 mM, KH2PO40.4 mM, NaHCO35 mM, glucose 5.5 mM, phenylmethylsulphonyl fluoride and a protease inhibitor cocktail (Sigma-Aldrich). To measure the enzymatic activity of Nox, 20 μL of homogenate was added to 80 μL Krebs buffer supplemented with 6.25 μM of lucigenin (Sigma-Aldrich). The reaction was initiated by the addition of 1 μL NADPH solution (Sigma-Aldrich) to a final concentration of 0.1 mM. Chemiluminescence counts were recorded using a Luminometer TD 20/20 (Turner Designs, Sunny-vale, CA, USA). The chemiluminescence was expressed in relative light units (RLU) per minute per mg protein.
Statistical analysis
All data are expressed as the mean ± SEM. Statistical analysis was evaluated using pairedt-test using SPSS 13.0 software (SPSS, Chicago, IL, USA). A value ofP< 0.05 was considered statistically significant.
Results
Nox2 protein expression within ischemic brain tissue after early-phase and late-phase reperfusion
The expression of Nox2 protein in the ischemic brain was detected after 90-minute MCAO followed by 3-hour (early phase) or 22.5-hour (late phase) reperfusion. Western blot analysis showed that non-ischemic brain tissue expressed low levels of Nox2 protein, while in ischemic brain tissue, Nox2 protein level was significantly increased after 22.5 hours of reperfusion (Figure 1A) (P< 0.05). This increase was not detected in the ischemic brain tissue after 3- hour reperfusion (Figure 1A). Nox2 protein band intensity was quantified and expressed as band intensity ratio of ischemic brain tissue to non-ischemic brain tissue (Figure 1B).
Nox2 mRNA expression within ischemic brain at early-phase and late-phase reperfusion
Nox2 mRNA expression in ischemic brain tissue was examined using real time quantitative RT-PCR. After 90-minute MCAO followed by 22.5-hour reperfusion, Nox2 mRNA expression was significantly increased in the ischemic brain tissue than in the non-ischemic brain tissue (P< 0.05;Figure 2). Similar to its protein results, Nox2 mRNA levels were comparable between non-ischemic and ischemic brain tissues after 3 hours of reperfusion (P> 0.05;Figure 2).
NADPH oxidase activity within ischemic brain at early-phase and late-phase reperfusion
NADPH oxidase activity was examined in the ischemic brain tissue after 90-minute MCAO, followed by 3 or 22.5 hours of reperfusion using lucigenin-enhanced chemiluminescence. As shown inFigure 3, NADPH oxidase activity was significantly increased in ischemic brain tissue than in non-ischemic brain tissue after 22.5 hours of reperfusion (P< 0.05), while no significant change was observed after 3 hours of reperfusion (P> 0.05), which was consistent with the results obtained above for Nox2 mRNA and protein expression.
Discussion
Restoration of blood flow to the ischemic brain is absolutely required for rescuing neuron-starving energy in ischemic stroke (Weaver and Liu, 2015). However, reperfusion is involved in massive ROS generation, which is believed to cause extra damage to the brain, resulting in post-ischemic hemorrhage, vasogenic brain edema, and neuronal death (Singhal, 2007; Rodrigo et al., 2013). Great efforts have been directed to the characterization of the sources of reperfusion-associated ROS generation within the brain (Bozkurt et al., 2014; Wu et al., 2015). Our results suggest that Nox2 may be an important enzymatic source of ROS generation at late-phase reperfusion, but may not contribute to reperfusion-associated early burst of ROS generation.
Nox2 is originally discovered in polymorphonuclear neutrophils and this enzyme is also expressed in many other non-phagocytic cells. In the past decades, the effect of Nox2 in ischemia/reperfusion brain damage has been increasingly appreciated as increased Nox2 expression is detected in ischemic brain tissue and inhibition of Nox2 by pharmacological inhibitors or genetic approaches significantly reduces cerebral infarct size and blood-brain barrier injuryin vivo(Liu et al., 2008; Kim et al., 2009; Thompson et al., 2012; Cooney et al., 2013; Kahles and Brandes, 2013; McCann et al., 2014). Of note, the majority of these studies have focused on Nox2 change in ischemic brain tissue following prolonged reperfusion. Similarly, results of this study showed that Nox2 mRNA and protein expression levels were upregulated in ischemicbrain tissue after 22.5 hours of reperfusion, and this upregulation was accompanied by an increase in NADPH oxidase activity. However, unlike prolonged reperfusion, the role of Nox2 in early reperfusion remains inconclusive. There are two recent studies showing that Nox2 is upregulated at 3 hours after reperfusion in ischemic stroke rats (Kochanski et al., 2013; Cai et al., 2016). However, in another study, McCann et al. (2014) demonstrated that Nox2 knockout had no effect on neuronal loss at 6 hours following post-ischemic reperfusion, while it reduced infarction volume at 24 hours after reperfusion onset (McCann et al., 2014). Our results showed that Nox2 was not upregulated at both transcriptional and translational levels at an early reperfusion time point (3 hours after reperfusion onset). This discrepancy may be due to different animal models used in these studies, which may induce different degrees of ischemia severity during the period of cerebral ischemia. Future studies are needed to test this possibility.
The Nox/Duox family of NADPH oxidases includes seven members, named as Nox1 to Nox5, dual oxidase (Duox)-1, and Duox-2 (Bedard et al., 2007). Although Nox2 has been considered the main isoform of NADPH oxidase relevant to ischemic stroke (Kahles and Brandes, 2013), Nox1, Nox4 and Duox-1 may also play a role in ischemic brain injury. Similar to Nox2, Nox4 appears to contribute to ROS generation in ischemic brain tissue following prolonged reperfusion as Nox4 mRNA expression is shown to be upregulated in ischemic brain tissue at 24 hours after MCAO onset, with an expression peak observed between days 7 and 15. Moreover, the peak phase of Nox4 mRNA expression coincides with the time of neoangiogenesis in the peri-infarct area, supporting a role of this enzyme in the restoration of ischemic brain damage (Vallet et al., 2005). In a rat model of transient MCAO, knockdown of Nox1 by adenovirus transfected Nox1 small hairpin RNA markedly reduces the injury size and attenuates neuron death, indicating an important role of Nox1 in ischemic brain damage (Choi et al., 2015). Duox1 upregulation has been observed in neurons, astrocytes and endothelial cells in a rat MCAO model, which may also contribute to ROS production in the ischemic brain tissue (Dvoriantchikova et al., 2012). This report only focused on the changes of Nox2 in ischemic brain tissue following reperfusion, and future studies are required to investigate other isoforms of NADPH oxidase to totally investigate the contribution of NADPH oxidases to ROS generation in the brain during post-ischemic reperfusion.
In conclusion, the present study demonstrated that Nox2 did not contribute to the burst of ROS generation in the ischemic brain at early-phase reperfusion, but was rather an important enzymatic source for ROS generation during prolonged reperfusion, which could help determine the timing of anti-NADPH oxidase treatments for acute ischemic stroke.
Author contributions:WLL and YZ conceived and designed the experiments. YZ, TW and KY performed the experiments. YJ, JX and JMW analyzed experimental data. YZ, TW and KY contributed to reagents/ materials/analysis tools. WLL wrote the paper. All authors approved the final version of this paper for publication.
Conflicts of interest:None declared.
Plagiarism check:This paper was screened twice using CrossCheck to verify originality before publication.
Peer review:This paper was double-blinded and stringently reviewed by international expert reviewers.
Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR (2005) Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25:9176-9184.
Amaro S, Chamorro A (2011) Translational stroke research of the combination of thrombolysis and antioxidant therapy. Stroke 42:1495-1499.
Babior BM (2004) NADPH oxidase. Curr Opin Immunol 16:42-47.
Bedard K, Lardy B, Krause KH (2007) NOX family NADPH oxidases: not just in mammals. Biochimie 89:1107-1112.
Beske PH, Jackson DA (2012) NADPH oxidase mediates the oxygen-glucose deprivation/reperfusion-induced increase in the tyrosine phosphorylation of the N-methyl-D-aspartate receptor NR2A subunit in retinoic acid differentiated SH-SY5Y Cells. J Mol Signal 7:15.
Bozkurt AA, Mustafa G, Tarık A, Adile O, Murat SH, Mesut K, Yıldıray K, Coskun S, Murat C (2014) Syringaldehyde exerts neuroprotective effect on cerebral ischemia injury in rats through anti-oxidative and anti-apoptotic properties. Neural Regen Res 9:1884-1890.
Cai L, Stevenson J, Geng X, Peng C, Ji X, Xin R, Rastogi R, Sy C, Rafols JA, Ding Y (2016) Combining normobaric oxygen with ethanol or hypothermia prevents brain damage from thromboembolic stroke via PKC-Akt-NOX modulation. Mol Neurobiol doi:10.1007/s12035-016-9695-7.
Chang CC, Wang YH, Chern CM, Liou KT, Hou YC, Peng YT, Shen YC (2011) Prodigiosin inhibits gp91(phox) and iNOS expression to protect mice against the oxidative/nitrosative brain injury induced by hypoxia-ischemia. Toxicol Appl Pharmacol 257:137-147.
Choi DH, Kim JH, Lee KH, Kim HY, Kim YS, Choi WS, Lee J (2015) Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS One 10:e0116814.
Cooney SJ, Bermudez-Sabogal SL, Byrnes KR (2013) Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation 10:155.
Dvoriantchikova G, Grant J, Santos AR, Hernandez E, Ivanov D (2012) Neuronal NAD(P)H oxidases contribute to ROS production and mediate RGC death after ischemia. Invest Ophthalmol Vis Sci 53:2823-2830.
Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D, Investigators E (2008) Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 359:1317-1329.
Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH (2010) Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol 41:172-179.
Kahles T, Brandes RP (2013) Which NADPH oxidase isoform is relevant for ischemic stroke? The case for nox 2. Antioxid Redox Signal 18:1400-1417.
Kim GS, Jung JE, Niizuma K, Chan PH (2009) CK2 is a novel negative regulator of NADPH oxidase and a neuroprotectant in mice after cerebral ischemia. J Neurosci 29:14779-14789.
Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, Janssen B, Gorlach A, Schmidt HH (2012) NADPH oxidases as a source of oxidative stress and molecular target in ischemia/ reperfusion injury. J Mol Med (Berl) 90:1391-1406.
Knox R, Brennan-Minnella AM, Lu F, Yang D, Nakazawa T, Yamamoto T, Swanson RA, Ferriero DM, Jiang X (2014) NR2B phosphorylation at tyrosine 1472 contributes to brain injury in a rodent model of neonatal hypoxia-ischemia. Stroke 45:3040-3047.
Kochanski R, Peng C, Higashida T, Geng X, Huttemann M, Guthikonda M, Ding Y (2013) Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: an inhibitory effect on hyperglycolysis and NADPH oxidase activation. J Neurochem 126:113-121.
Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK (2003) Honokiol protects rat brain from focal cerebral ischemia-reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res 992:159-166.
Liu J, Jin X, Liu KJ, Liu W (2012) Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci 32:3044-3057.
Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ (2004) Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 24:343-349.
Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, Miyake M, Liu KJ (2008) Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem 107:1196-1205.
Manea A, Manea SA, Gan AM, Constantin A, Fenyo IM, Raicu M, Muresian H, Simionescu M (2015) Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem Biophys Res Commun 461:172-179.
McCann SK, Dusting GJ, Roulston CL (2014) Nox2 knockout delays infarct progression and increases vascular recovery through angiogenesis in mice following ischaemic stroke with reperfusion. PLoS One 9:e110602.
Ono T, Tsuruta R, Fujita M, Aki HS, Kutsuna S, Kawamura Y, Wakatsuki J, Aoki T, Kobayashi C, Kasaoka S, Maruyama I, Yuasa M, Maekawa T (2009) Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res 1305:158-167.
Ostrowski RP, Tang J, Zhang JH (2006) Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke 37:1314-1318.
Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC, Goldschmidt-Clermont PJ, Irani K (2000) Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J 14:418-429.
Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12:698-714.
Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL (2015) Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia 63:466-482.
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M (2013) Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47:9-23.
Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E (2003) NADPH oxidase immunoreactivity in the mouse brain. Brain Res 988:193-198.
Singhal AB (2007) A review of oxygen therapy in ischemic stroke. Neurol Res 29:173-183.
Tang X, Zhong W, Tu Q, Ding B (2014) NADPH oxidase mediates the expression of MMP-9 in cerebral tissue after ischemia-reperfusion damage. Neurol Res 36:118-125.
Thompson JW, Narayanan SV, Perez-Pinzon MA (2012) Redox signaling pathways involved in neuronal ischemic preconditioning. Curr Neuropharmacol 10:354-369.
Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I (2005) Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 132:233-238.
Weaver J, Liu KJ (2015) Does normobaric hyperoxia increase oxidative stress in acute ischemic stroke? A critical review of the literature. Med Gas Res 5:11.
Wu JX, Zhang LY, Chen YL, Yu SS, Zhao Y, Zhao J (2015) Curcumin pretreatment and post-treatment both improve the antioxidative ability of neurons with oxygen-glucose deprivation. Neural Regen Res 10:481-489.
Copyedited by Wang J, Li CH, Song LP, Zhao M
*Correspondence to: Wen-lan Liu, M.D., Ph.D., wlliu@szu.edu.cn.
orcid: 0000-0003-2130-4065 (Wen-lan Liu)
10.4103/1673-5374.194747
Accepted: 2016-09-20

- 中国神经再生研究(英文版)的其它文章
- The status of Nrf2-based therapeutics: current perspectives and future prospects
- Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders
- Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification
- Applicability of tooth derived stem cells in neural regeneration
- Cortical spreading depression-induced preconditioning in the brain
- Neuroinflammation, neurodegeneration and regeneration in multiple sclerosis: intercorrelated manifestations of the immune response