
保留型与反转型β-木糖苷酶活性位点的分子动力学模拟
2015-12-29 02:33:27张俊威周峻岗
物理化学学报 2015年6期
张俊威 周峻岗 吕 红 黄 强
(1复旦大学生命科学学院,遗传工程国家重点实验室,上海工业菌株工程技术研究中心,上海200433;2上海生物制造技术协同创新中心,上海200237)
保留型与反转型β-木糖苷酶活性位点的分子动力学模拟
张俊威1周峻岗1,*吕 红1,2黄 强1,2,*
(1复旦大学生命科学学院,遗传工程国家重点实验室,上海工业菌株工程技术研究中心,上海200433;2上海生物制造技术协同创新中心,上海200237)
木聚糖是潜在的重要可再生能源,如何提高其降解效率已成为近年来的研究热点.β-木糖苷酶是木聚糖降解过程中的关键酶之一,按其水解机制可分为保留型与反转型酶.目前虽然对于这两种催化机制的研究不断深入,但很少有工作从溶液环境的角度出发探究它们的差异.本文采用分子动力学模拟方法,对4个典型的β-木糖苷酶进行了显式溶剂模拟研究,详细分析了酶的催化氨基酸间的距离和质子供体氨基酸p Ka值的动态变化.结果显示,反转型酶催化氨基酸间的距离约为0.8-1.0 nm,大于保留型的0.5-0.6 nm,与先前对糖苷酶晶体结构的统计分析结果一致.令人意外的是,保留型酶的质子供体通过与其附近组氨酸的相互作用,其p Ka在两个不同的高、低值之间交替变换,使保留型酶的双取代反应得以发生;而反转型酶的质子供体则由附近的天冬氨酸调节,其p Ka稳定在某个较高值,这可能有利于其在反应pH值下获得水溶液中的氢离子,进行反转型酶特有的单取代反应.因此,本工作加深了人们对β-木糖苷酶保留型与反转型水解机制的认识,并为后续酶的理性改造与高效利用提供具有指导价值的结构与机理信息.
β-木糖苷酶;催化机制;分子建模;质子供体;p Ka值
1 引言
随着石油等化石类能源的不断减少,生物质能源逐渐受到了人们的关注.虽然木质纤维素等生物能源具有再生性、清洁低碳及原料丰富等优势,但是目前生物质的利用率还很低.因此,如何高效地利用生物质能源已成为一个急需解决的生物技术问题和当前的研究热点.1,2
木聚糖作为半纤维素的主要组成部分,3是重要的待开发的生物质能源.β-木糖苷酶是木聚糖降解过程中关键酶之一,分布广泛,主要存在于植物、真菌和细菌中.4,5该酶以外切方式作用于木寡糖和木二糖的β-1,4-糖苷键,可减缓低聚木糖等中间产物对内切木聚糖酶的抑制作用,6进而加快木聚糖的整体降解效率.根据水解产物的异头碳构型在反应前后是否保留或反转,可将β-木糖苷酶的催化机制分为保留型和反转型.7已发现的β-木糖苷酶大多采用保留型机制,分布于糖苷水解酶GH3、GH39、GH52和GH54家族;而少数β-木糖苷酶为反转型酶,仅存在于GH8和GH43家族(http://www.cazy.org).
目前人们已经比较清楚:β-木糖苷酶的水解过程为一般意义上的酸碱催化反应,活性位点处需要两个关键的催化氨基酸,一个作为质子供体,另一个作为亲核试剂.8,9其中,保留型酶采用双取代反应机制:10首先由质子供体为糖苷键氧原子提供一个质子,促使糖苷键断裂,接着亲核试剂进攻糖基的异头碳,形成稳定中间体,最后质子供体协助进入活性中心的水分子进攻糖基的碳正离子,完成糖基取代反应(图1a);反转型酶则采用单取代机制:11先由质子供体为底物糖苷键的氧原子提供一个质子,同时亲核试剂与活性中心的水分子反应,使水分子解离产生羟基,并与糖苷键的碳正离子结合,从而打开糖苷键(图1b).
在上述水解过程中,保留型酶较易发生转糖基作用,因此其水解效率不如反转型酶的效率高.12人们推测一个主要原因可能是保留型酶的水解过程需越过两个反应过渡态(图1a),而反转型酶只有一个过渡态(图1b).总的说来,对于决定β-木糖苷酶水解效率的关键因素,人们还是知之甚少.因此,深入分析上述两种水解机制的差异,揭示影响水解效率的主要因素,对β-木糖苷酶的开发与应用具有重要意义.已有一些研究工作对两种催化机制间的差别进行了一定的探讨,13例如,比较分析了多种糖苷酶晶体结构中催化氨基酸之间的距离,14以及它们的p Ka值15,16等.不过,这些工作基本上是以酶的静态结构为分析基础的,关于在溶液环境中酶活性位点动态变化的分析还很少见报道.所以,本文应用分子动力学(MD)模拟方法对溶液环境中β-木糖苷酶活性位点的动态行为进行研究.分子动力学模拟作为实验方法的补充,能够详细了解溶液中生物大分子的微观结构、热力学和动力学性质等相关信息,17从而在微观层面解释蛋白质的性质和功能.18本文以保留型与反转型β-木糖苷酶为对象,通过显式溶剂的分子动力学模拟,分析酶活性位点中催化氨基酸间的距离、质子供体氨基酸p Ka的动态变化,从而深入理解β-木糖苷酶保留型与反转型水解机制间的差异,为后续的酶的理性改造和高效利用提供了具有指导意义的信息.
2 计算方法
2.1 MD模拟体系的选择与构建
在本工作开始时,我们仔细检索了蛋白质结构数据库(Protein Data Bank,http://www.rcsb.org),发现具有完整晶体结构的β-木糖苷酶共有8个.其中, GH39家族的两个酶(PDB codes:1PX8,194EKJ20)和GH43家族的两个酶(PDB codes:2EXH,213C2U22)的催化机制已较为明确,且分别代表了保留型与反转型酶,其详细信息见表1.因此,本工作选取这4个晶体结构建立MD模拟体系.
利用上述晶体结构,使用GROMACS(版本: 4.6.5)软件包23的pdb2gmx模块构建了4个β-木糖苷酶的全原子模型.为了进一步了解酶催化氨基酸p Ka值的变化情况,对表1中的质子供体氨基酸进行质子化与非质子化处理,以模拟该残基两种不同的质子化状态.这样,为表1中的每一个β-木糖苷酶各构建了2个MD模拟体系.对每个体系,都采用Amber99sb力场24描述其原子的各种参数,并按全原子显式溶剂方法构建:首先把酶置于立方盒子中央,使其离盒子边界至少1.2 nm,然后往盒子中加入水分子,水分子模型为TIP3P,25最后加入一定数目的钠离子(Na+)与氯离子(Cl-),使体系的离子浓度值为150mmol·L-1,且体系保持中性,总电荷数为零.

图1 保留型(a)和反转型(b)β-木糖苷酶的催化机制Fig.1 Catalyticmechanismsof retaining(a)and inverting(b)β-xylosidases
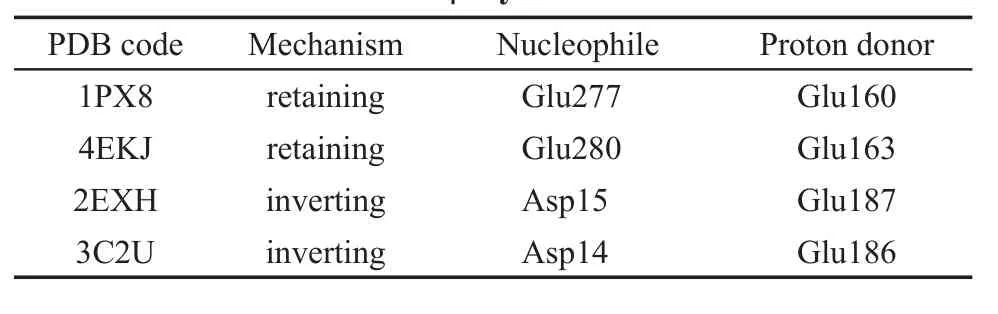
表1 4个β-木糖苷酶的催化机理及催化氨基酸Table1 Catalyticmechanismsand catalytic am ino-acids of fourβ-xylosidases
2.2 分子动力学模拟
采用GROMACS的mdrun模块进行MD模拟.模拟开始前,用最速下降法对体系进行5000步能量最小化,消除体系内可能存在的不合理的原子位置或接触.然后,对体系进行预平衡模拟:在约束蛋白质重原子位置的情况下,进行100 ps的恒温恒容(NVT)模拟,使体系温度达到并保持在300 K;然后,在恒温恒压(NPT)条件下,继续对体系平衡1000 ps,使体系的压强保持在105Pa.预平衡模拟结束后,不再约束重原子的位置,接着进行时间尺度为20 ns的NPT模拟,产生用于后续分析的MD模拟轨迹.在模拟中,积分步长为2 fs,长程静电相互作用采用particle-mesh Ewald(PME)方法26计算,用Berendsen弱耦合作用27稳定体系的温度和压强,耦合时间常数为0.1 ps.模拟过程中,每2 ps保存一次体系原子的坐标与速度,以用于后续分析.模拟轨迹的显示与构象分析主要用VMD(版本:1.9.1)28与PYMOL(版本:1.4.1)29完成.
2.3 MD模拟轨迹分析
2.3.1 催化氨基酸间距离的计算
在水解反应发生时,β-木糖苷酶的质子供体氨基酸带有一个质子(图1).为了解在水解反应的条件下酶的两个催化氨基酸之间的距离,我们按2 ps的间隔从相应酶体系的MD模拟轨迹中提取酶原子的位置坐标,生成相应的PDB文件,然后使用与文献30类似的方法计算β-木糖苷酶的两个催化残基之间的距离.首先,按图2的方式定义两个催化氨基酸羧基氧原子的位置,然后用距离公式d=[(x1-x2)+(y1-y2)+ (z1-z2)]1/2分别计算酶Oδ1或Oδ2到Oε1、Oε2之间的距离(即d11、d12、d21和d22),再取这四个值的平均值得到表征催化氨基酸之间距离的统计值Doo=(d11+d12+d21+ d22)/4.
2.3.2 质子供体氨基酸p Ka值的计算
在一定的pH条件下,蛋白质中酸性与碱性氨基酸可根据其自身的p Ka值来调节其质子化状态,31从而影响蛋白质的功能.32由于β-木糖苷酶的质子供体氨基酸在反应pH值下需带上一个质子,水解反应才能发生(图1).因此,分析该氨基酸残基p Ka值的动态变化,可以了解其在反应pH值下是否带有水解反应所必需的质子.如上所述,为准确计算该氨基酸残基p Ka值,我们构建了4个相应的、质子供体氨基酸不带质子的酶模拟体系.同样,按2 ps的间隔从这些酶体系的模拟轨迹中提取酶原子的位置坐标,生成PDB文件后,用PROPKA(版本:3.1)33分别计算出各PDB文件中质子供体氨基酸的p Ka值.
3 结果与讨论
3.1 酶氨基酸残基的波动分析
为判断各模拟体系到达平衡的模拟时间,用GROMACS的g_rms模块分别计算上述8个体系中酶的Cα原子相对于初始结构的均方根偏差(RMSD),结果如图3所示.可以看出,各个体系经5 ns模拟后Cα原子的RMSD值已基本趋于稳定,说明各体系的MD模拟已达到了平衡.所以,后续各体系的分析使用的是其5-20 ns的模拟轨迹.
为了了解模拟过程中酶的局部运动情况,用GROMACS的g_rmsf模块对酶催化结构域中的每一个氨基酸残基进行均方根波动(RMSF)分析,结果见图4.从图4a、4b中的RMSF曲线来看,无论是保留型还是反转型酶,即使是活性中心无底物存在情况下,酶的两个催化氨基酸基本上是稳定的,热运动的幅度相对较小.在模拟过程中,运动幅度相对较大的是酶结构外围的氨基酸残基,特别是非规则的环状链段,如图4(c-f)中的MD构象所示.

图2 催化氨基酸残基间距离的定义Fig.2 Definition of the distancesbetween am ino-acid residues
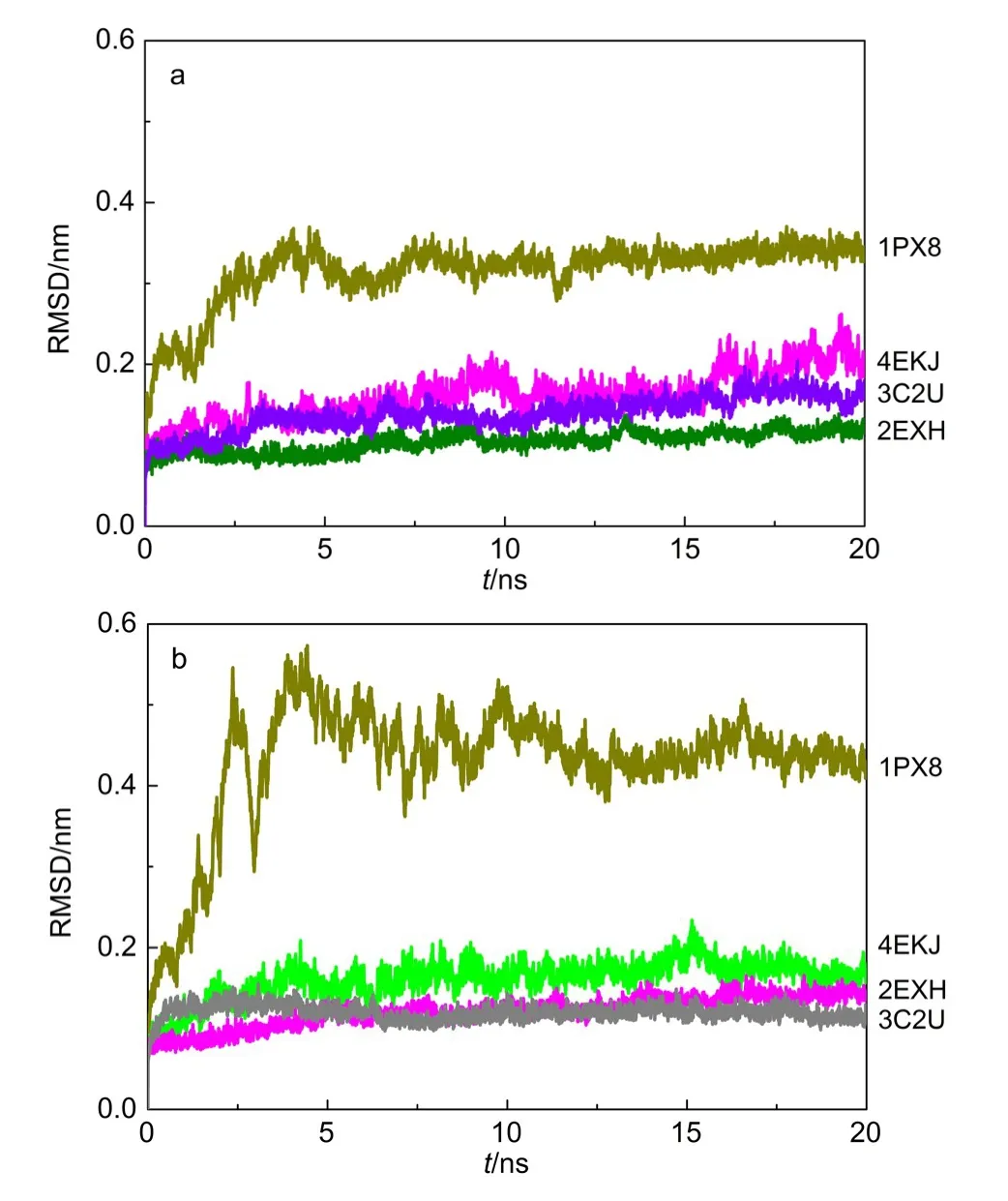
图3 8个模拟体系Cα碳原子的RMSD曲线Fig.3 Rootmean square deviation(RMSD)curvesof Cαatom s for 8 simulated system s
3.2 催化氨基酸残基间距离的分析
采用表1中质子供体氨基酸质子化的4个酶体系的模拟轨迹,根据上述距离Doo的计算式分别计算了保留型和反转型酶的催化氨基酸间距离Doo随模拟时间变化的情况及其概率分布(图5).由图可见,随着模拟时间的增加,Doo趋向于稳定值,但由于受到溶液环境中水分子的影响,Doo有小幅度的波动.利用模拟中波动较小的5-20 ns的轨迹,计算了Doo的平均值及其标准偏差:保留型酶1PX8中两个催化残基间的距离分布基本保持在(0.58±0.04)nm(图5a),4EKJ则在(0.59±0.04)nm附近上下波动(图5b);反转型酶2EXH的距离为(0.81±0.03)nm(图5c), 3C2U为(1.05±0.05)nm(图5d).
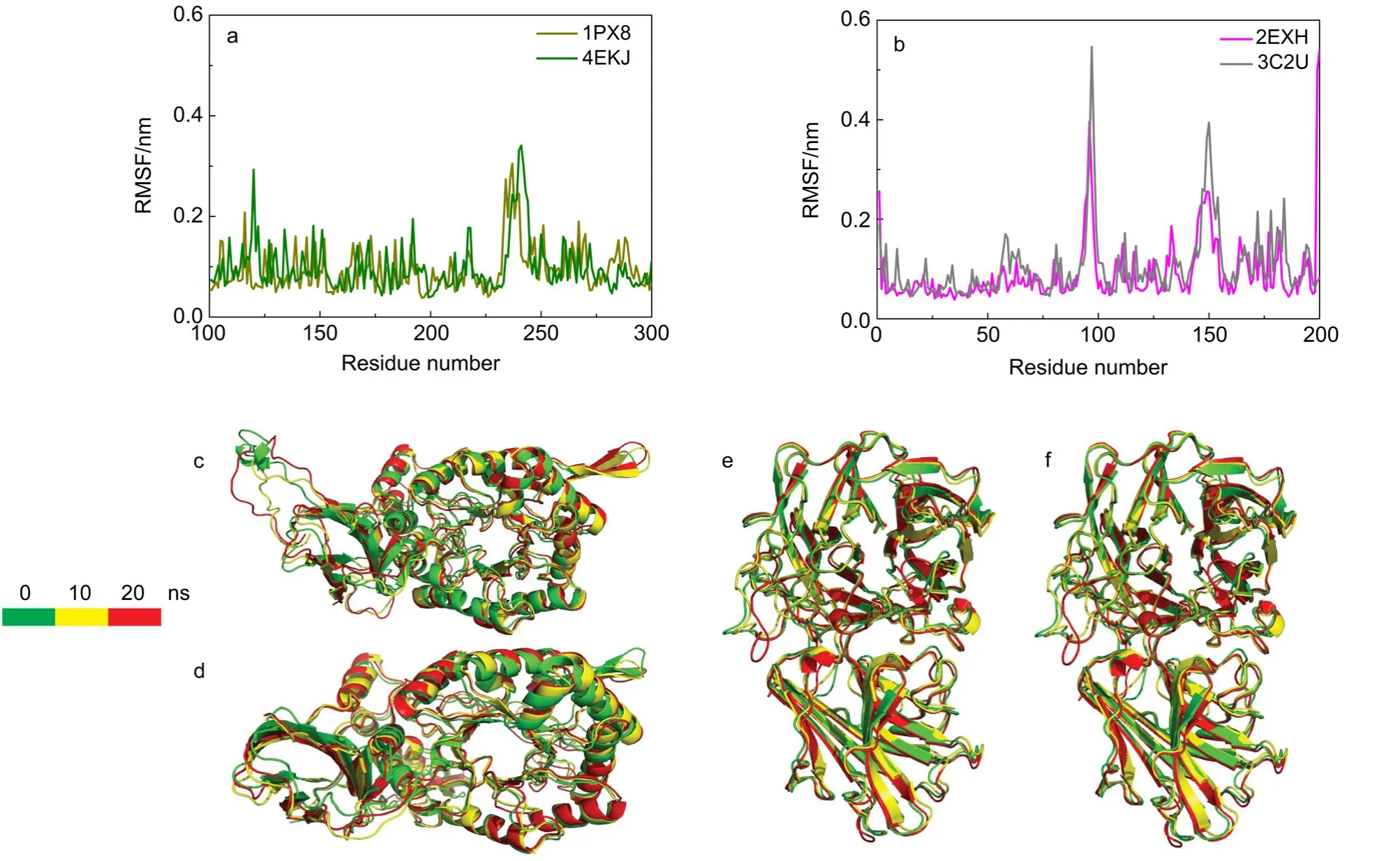
图4催化结构域氨基酸残基RM SFs及MD构象Fig.4 Rootmean square fluctuations(RM SFs)of am ino-acidsand MD conformations for the catalytic dom ains
上述结果说明,反转型酶的两个催化氨基酸之间的距离比保留型酶的距离要大,主要原因是反转型酶的催化过程中有水分子的直接参与,需要较大的空间同时容纳底物与水分子.这个结果也验证了以前的发现:反转型酶催化氨基酸间的距离较远(~1.0 nm),而保留型酶的距离则相对较小(~0.55 nm).14不过,Mhlongo等30对36个糖苷酶家族共136个晶体结构进行统计分析后发现:由于家族的不同,保留型酶催化氨基酸间的距离一般为(0.48± 0.03)nm或者(0.64±0.06)nm,不完全等于0.55 nm;而反转型酶的距离变化范围较宽泛((0.8±0.2)nm),也不完全等于1.0 nm.本工作的结果与此相一致,同时也直接说明了溶液环境中催化残基之间的距离基本稳定.不难理解,这种稳定的运动方式有利于底物以特定的模式结合到酶的活性中心,并形成后续反应的过渡态,从而高效地催化糖苷键的水解反应.
为进一步了解距离差异产生的原因,我们对这2类β-木糖苷酶的三维结构进行了比较,同时采用wordom程序34统计了催化氨基酸附近的水分子数,利用VMD追踪观察出现几率最大的水分子在催化氨基酸附近的运动情况.从图6可以看出,保留型酶1PX8具有典型的(β/α)8桶状结构,即分别由8个β折叠和8个α螺旋构成;而反转型酶2EXH则是由5组β折叠组成催化结构域.两者的催化结构域的拓扑结构都类似口袋,且其深度限定两者的水解方式为外切型.8然而,催化氨基酸附近水分子的运动方式十分不同:1PX8的两个催化残基处于活性口袋的表面,且距离较小,此时水分子较为集中在质子供体(Glu160)附近(图6a),只有当质子供体为反应提供质子后,才能对其进行活化,完成双取代反应;而在2EXH中,质子供体(Glu187)处于口袋表面,亲核试剂(Asp15)在口袋底部,这种排布方式增加了两个残基间的距离,为水分子在口袋内提供了驻留空间,使其较为集中在Asp15附近(图6b),有利于直接参与水解反应.
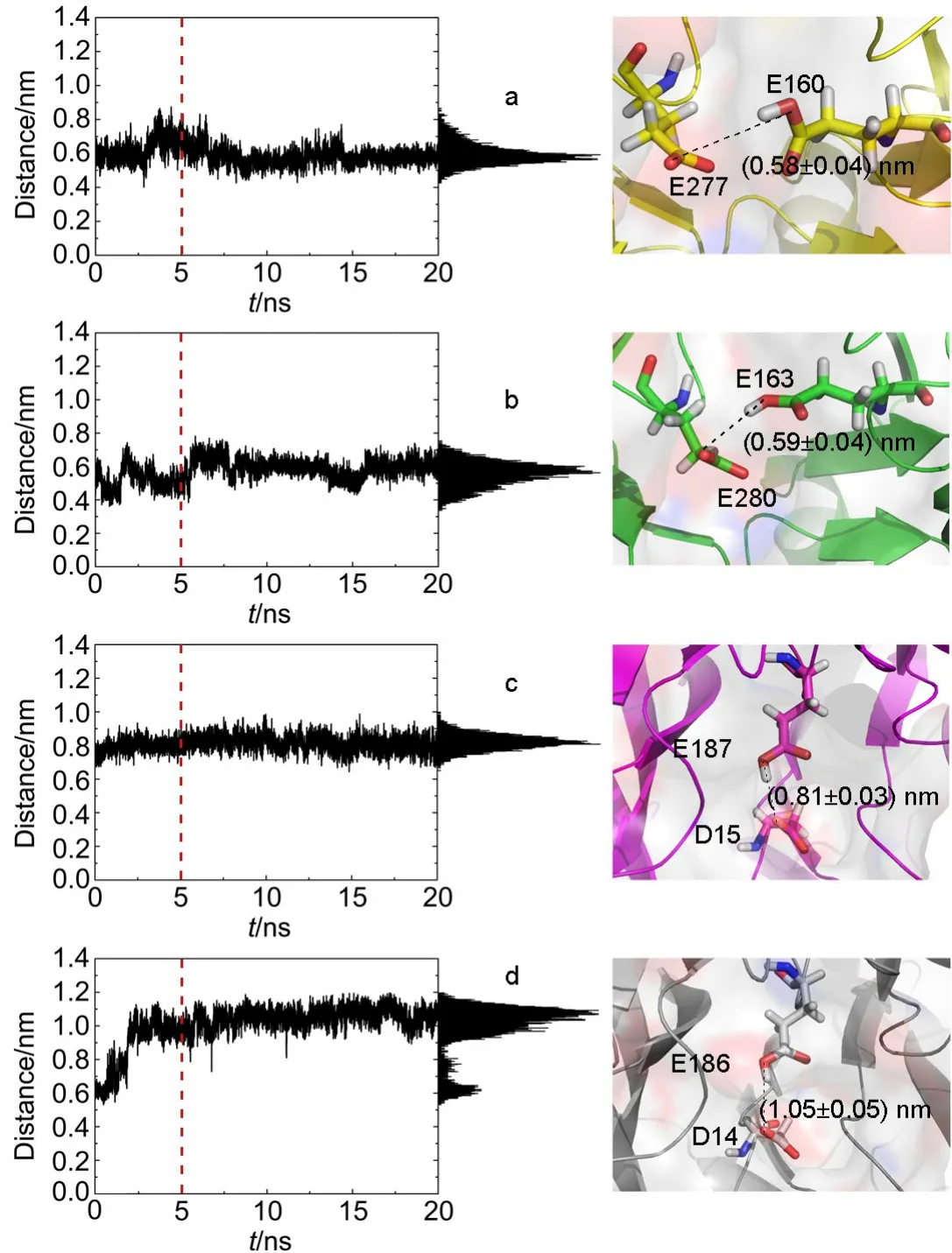
图5 活性口袋中质子供体与亲核试剂之间距离随模拟时间的变化(左),概率分布(中)和距离结构示意图(右)Fig.5 Distance changesbetween the proton donorsand the nucleophiles in active-site pocketsw ith simulated time(left),their probability distributions(m idd le),and structuraldiagram s(right)
3.3 质子供体p Ka值的分析
除统计分析催化氨基酸之间的距离外,本工作还着重探究了酶质子供体(谷氨酸)p Ka在MD模拟中的动态变化情况.利用p Ka值计算程序PROPKA(见计算方法),计算了各个酶模拟轨迹中的质子供体p Ka值随模拟时间的变化及概率分布,结果如图7所示.由图可见,在apo的状态下,受活性位点的微环境的影响,各酶体系中质子供体p Ka值都偏离了氨基酸的本征值(4.3).一个非常令人意外的发现是,在概率分布图上,两个保留型酶(1PX8和4EKJ)都出现了两个峰值,一个值较大,另一个较小(图7(a,b));而两个反转型酶(2EXH和3C2U)则分别围绕不同的定值上下波动(图7(c,d)).因此,我们推断,β-木糖苷酶的催化机制不仅与活性口袋的大小有关,同时也受到质子供体p Ka值的影响.
为了分析保留型和反转型酶的质子供体p Ka值之间的具体差异,同时进一步理解溶液pH对于催化效率的影响,我们利用5-20 ns的模拟轨迹,计算了p Ka的平均值及其标准偏差,结果列于表2.以往的研究已经发现,不同的β-木糖苷酶水解反应的最适pH值各不相同,但多为酸性.35,36从图1的催化机制可见,反应开始前β-木糖苷酶的质子供体带有H+,因此,处于活性中心的质子供体的p Ka值需要大于溶液环境pH值,以保证在反应pH值下质子供体带上一个H+,具备催化水解反应的能力.由表2可以看出,反应开始前各体系质子供体基本处于弱碱性状态.这与前面的设想吻合,即质子供体的p Ka值一定程度上会决定了其反应最适pH值.
由于木糖苷酶的催化反应十分迅速,实验上难于获得酶-底物复合物的晶体结构,模拟底物存在情况下酶活性位点的动态性质还有一定的难度,因为不易确定底物分子在活性位点处的初始构象.不过,为进一步了解当底物分子进入活性中心后质子供体p Ka值的变化情况,我们以上述4个蛋白质中唯一含有底物的复合物晶体结构2EXJ(2EXH的突变体:D128G)为研究对象,利用MODELLER37构建野生型酶-底物的复合物结构作为MD模拟的初始构象.在此,根据无底物分子(apo)状态下质子供体(E187)的p Ka值为7.8,大于环境的pH值,对其进行质子化处理,然后用2.2节中的方法进行MD模拟,其中底物木二糖分子采用GLYCAM 06力场,38并统计模拟轨迹(5-20 ns)的质子供体p Ka值,结果如图8所示.由图中可见,当木二糖结合到活性口袋时,质子供体由于受到底物分子的影响,其p Ka值由apo状态的7.8±0.4降到1.5±0.2,小于反应溶液的pH值,此时残基易失去质子,并传递给木二糖,从而完成催化反应.可见,上述apo的结果已清晰地反映了保留型与反转型酶质子供体p Ka值的动态差异.

图6 保留型酶(a)与反转型酶(b)的结构及活性口袋内水分子扩散比较Fig.6 Com parisonsof the structuresand water diffusions in active-site pocketsof retaining(a) and inverting(b)enzymes

图7 质子供体p Ka值随模拟时间的变化及其概率分布Fig.7 p Kavalue changesof the proton donorsw ith simulated time and their probability distributions

表2 各研究体系中质子供体p Ka平均值及其标准偏差Table2 M ean p Kavaluesand the standard deviations of proton donorsof all the studied system s
3.4 保留型酶突变体中质子供体p Ka值的分析
对反转型酶2EXH的研究曾发现,质子供体Glu187的p Ka值可以增加到7.1,但需要其附近天冬氨酸Asp128来调节并稳定其p Ka值.39根据此结论,我们也对保留型酶1PX8的结构进行分析,发现质子供体Glu160附近存在一个保守的单质子化的组氨酸His228(图6),对质子供体的p Ka值有影响:当其靠近Glu160时,p Ka值降低,反之,p Ka值升高.因此,利用MODELLER分别构建了保留型酶1PX8 (H228A)和4EKJ(H229A)的突变体,并进行了与野生型酶完全类似的MD模拟,计算质子供体的p Ka值,结果如图9所示.由图中可见,当1PX8的His228突变为Ala(丙氨酸)时,其质子供体p Ka值的两个峰都往高处移动了,而4EKJ(H229A)的两个峰则逐渐向中间靠拢,变为一个单峰.由此可见,保守氨基酸His对于保留型催化机制质子供体p Ka值的动态变化起着关键的调节作用.而且,很有可能可以通过改变His位点的氨基酸类型来转变酶的催化机制.
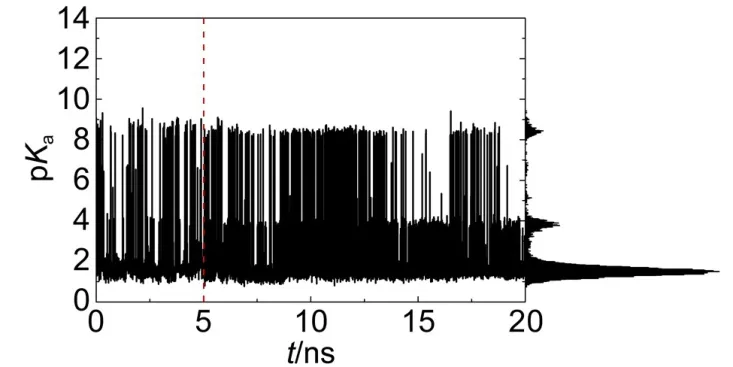
图8 含木二糖的2EXH中质子供体p Ka值随模拟时间的变化及其概率分布Fig.8 Changes in p Kavalue of the proton donors in 2EXH w ith xylobiosew ith simulated timeand their probability distributions
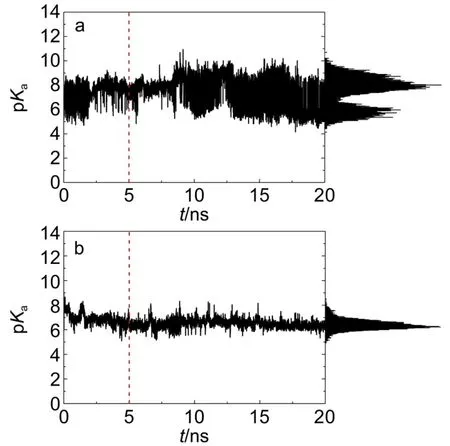
图9 保留型酶突变体中质子供体p Ka值随模拟时间的变化及其概率分布Fig.9 Changes in p Kavaluesof the proton donors in them utantsof retaining system sw ith simulated timeand their probability distributions
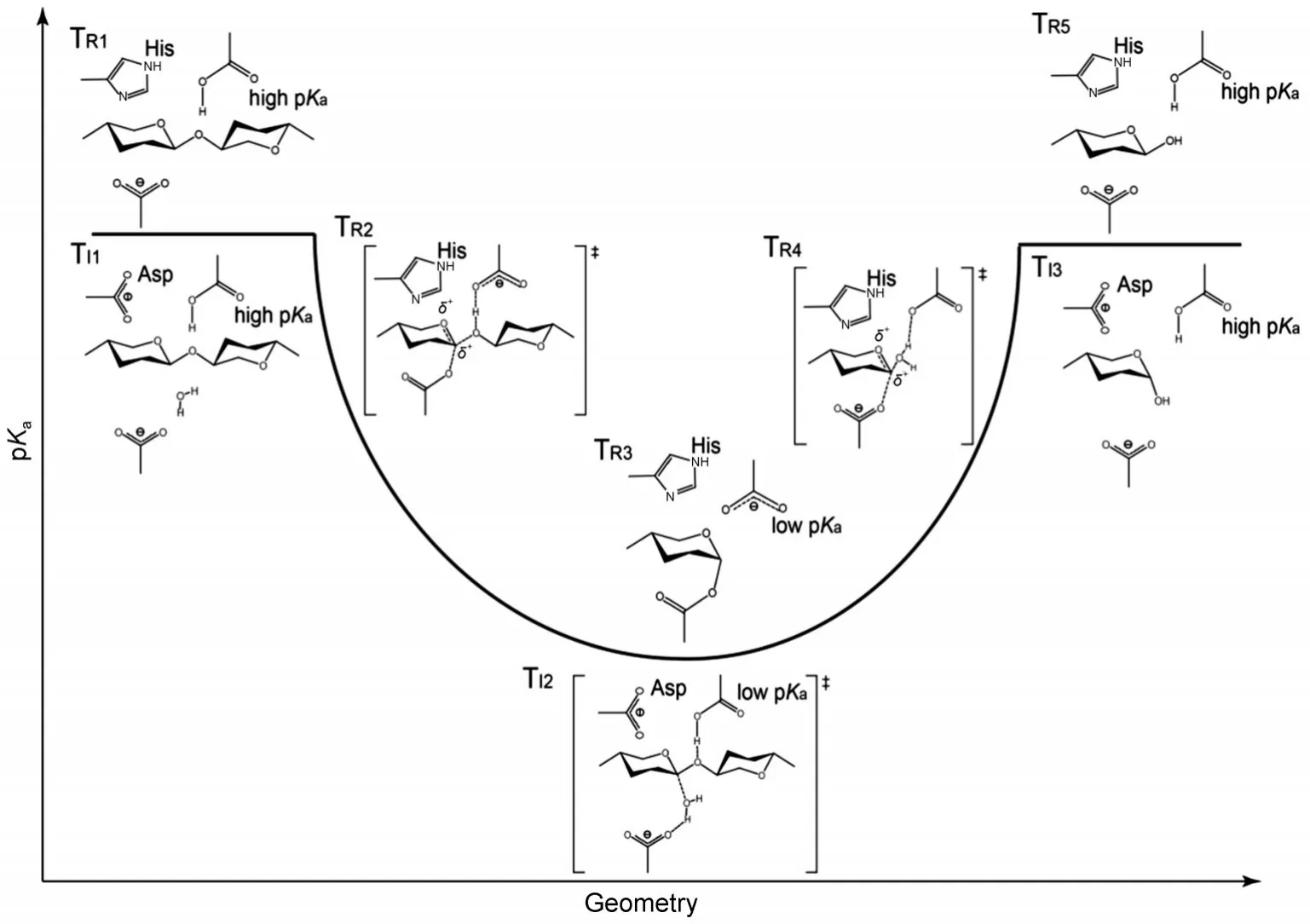
图10 酶催化过程中质子供体p Ka值的变化示意图Fig.10 Schematic diagram for the p Kavalue changesof proton donors in the enzymatic catalysis
3.5 木糖苷酶的催化机制与质子供体p Ka值的关系
为解释质子供体p Ka值是如何影响β-木糖苷酶的催化机制,基于上述结果我们提出了如图10所示的质子供体p Ka影响催化机制的模式.在保留型催化机制中,质子供体的p Ka值稳定在两个不同的值,以满足双取代反应过程中对其质子化状态的要求(如中间状态TR1、TR3):10,40当其p Ka值较高时,TR1可以充当质子供体,为糖苷键氧原子提供一个质子,而当其p Ka值较低时,TR3可以对进入活性中心的水分子进行去质子化.在反转型催化机制中,需稳定在某个较高值(质子化中间状态TI1),这样有利于其获得水溶液中的H+;而当底物进入活性中心时(TI2),质子供体的p Ka值则降至小于环境pH的较低值,为后续的反应提供质子,完成单取代反应.但在此过程中,质子供体p Ka值的动态变化主要是受到附近其它保守残基的影响:在反转型催化机制中,其质子供体的高p Ka值需要附近的天冬氨酸来调节,稳定其p Ka值;39而保留型机制中,组氨酸通过调节其与质子供体间的距离,从而使质子供体的p Ka值在反应过程中发生高低交替变化.
4 结论
本文采用MD模拟方法研究了溶液环境下保留型与反转型β-木糖苷酶活性位点动态行为的差异.通过对4个典型的β-木糖苷酶的分析,获得了反转型酶催化残基间的距离大于保留型酶残基间距离的结论,与前人的发现相一致.令人意外的是,本工作发现:保留型酶的质子供体的p Ka在高低两个定值之间交替变化,这也是保留型酶双取代反应得以发生的基本要求;而反转型中质子供体的p Ka则稳定在某个较高值,以利于获得水溶液中的氢离子,进行单取代反应.另外,我们还发现质子供体附近存在保守氨基酸,如组氨酸、天冬氨酸,可以通过原子间的相互作用来调节质子供体p Ka值的动态变化,从而有可能通过改变这些位点的氨基酸来最终转变酶的水解机制.这些结果进一步阐明了保留型与反转型β-木糖苷酶催化机制间的差异,加深了对β-木糖苷酶水解机制的认识,为后续酶的理性改造和高效利用提供具有指导价值的结构与机理信息.
(1)Himmel,M.E.;Ding,S.Y.;Johnson,D.K.;Adney,W.S.; Nim los,M.R.;Brady,J.W.;Foust,T.D.Science 2007,315 (5813),804.doi:10.1126/science.1137016
(2)Dionisi,D.;Anderson,J.A.;Aulenta,F.;M cCue,A.;Paton,G. J.Chem.Technol.Biotechol.2014,90(3),366.doi:10.1002/ jctb.4544
(3)Shallom,D.;Shoham,Y.Curr.Opin.M icrobiol.2003,6(3), 219.doi:10.1016/s1369-5274(03)00056-0
(4)Lorenz,W.W.;Wiegel,J.J.Bacteriol.1997,179(17),5436.
(5)M inic,Z.;Rihouey,C.;Do,C.T.;Lerouge,P.;Jouanin,L. Plant.Physiol.2004,135(2),867.doi:10.1104/pp.104.041269
(6)Sunna,A.;Antranikian,G.Crit.Rev.Biotechnol.1997,17(1), 39.doi:10.3109/07388559709146606
(7)Knob,A.;Terrasan,C.R.F.;Carmona,E.C.World.J.M icrob. Biot.2010,26(3),389.doi:10.1007/s11274-009-0190-4
(8)Davies,G.;Henrissat,B.Structure 1995,3(9),853.doi: 10.1016/S0969-2126(01)00220-9
(9)Rye,C.S.;Withers,S.G.Curr.Opin.Chem.Biol.2000,4(5), 573.doi:10.1016/S1367-5931(00)00135-6
(10)White,A.;Rose,D.R.Curr.Opin.Struc.Biol.1997,7(5),645. doi:10.1016/s0959-440x(97)80073-5
(11)Barker,I.J.;Petersen,L.;Reilly,P.J.J.Phys.Chem.B 2010, 114(46),15389.doi:10.1021/jp107886e
(12)Jordan,D.B.;Wagschal,K.;Grigorescu,A.A.;Braker,J.D. Appl.Microbiol.Biotechnol.2013,97(10),4415.doi:10.1007/ s00253-012-4475-4
(13)Vasella,A.;Davies,G.J.;Bohm,M.Curr.Opin.Chem.Biol. 2002,6(5),619.doi:10.1016/s1367-5931(02)00380-0
(14)M ccarter,J.D.;Withers,S.G.Curr.Opin.Struc.Biol.1994,4 (6),885.doi:10.1016/0959-440x(94)90271-2
(15)Zechel,D.L.;Withers,S.G.Curr.Opin.Chem.Biol.2001,5 (6),643.doi:10.1016/s1367-5931(01)00260-5
(16)Ludw iczek,M.L.;D'Angelo,I.;Yalloway,G.N.;Brockerman, J.A.;Okon,M.;Nielsen,J.E.;Strynadka,N.C.J.;Withers,S. G.;M cIntosh,L.P.Biochemistry 2013,52(18),3138.doi: 10.1021/bi400034m
(17)Dong,X.Y.;Du,W.J.;Liu,F.F.Acta Phys.-Chim.Sin.2012, 28,2735.[董晓燕,都文婕,刘夫锋.物理化学学报,2012, 28,2735.]doi:10.3866/pku.whxb201207162
(18)Dror,R.O.;Dirks,R.M.;Grossman,J.P.;Xu,H.F.;Shaw,D. E.Ann.Rev.Biophys.2012,41,429.doi:10.1146/annurevbiophys-042910-155245
(19)Yang,J.K.;Yoon,H.J.;Ahn,H.J.;Lee,B.I.;Pedelacq,J.D.; Liong,E.C.;Berendzen,J.;Laivenieks,M.;Vieille,C.;Zeikus, G.J.;Vocadlo,D.J.;Withers,S.G.;Suh,S.W.J.Mol.Biol. 2004,335(1),155.doi:10.1016/j.jmb.2003.10.026
(20)Santos,C.R.;Polo,C.C.;Correa,J.M.;Simao,R.D.G.; Seixas,F.A.V.;Murakam i,M.T.Acta Crystallogra.D-Biol. Crystallogr.2012,68(Suppl.10),1339.doi:10.1107/ s0907444912028491
(21)Brux,C.;Ben-David,A.;Shallom-Shezifi,D.;Leon,M.; Niefind,K.;Shoham,G.;Shoham,Y.;Schomburg,D.J.Mol. Biol.2006,359(1),97.doi:10.1016/j.jmb.2006.03.005
(22)Brunzelle,J.S.;Jordan,D.B.;M cCaslin,D.R.;Olczak,A.; Waw rzak,Z.Arch.Biochem.Biophys.2008,474(1),157.doi: 10.1016/j.abb.2008.03.007
(23)Hess,B.;Kutzner,C.;van der Spoel,D.;Lindahl,E.J.Chem. Theory Comput.2008,4(3),435.doi:10.1021/ct700301q
(24)Hornak,V.;Abel,R.;Okur,A.;Strockbine,B.;Roitberg,A.; Simmerling,C.Proteins2006,65(3),712.doi:10.1002/ prot.21123
(25)Jorgensen,W.L.;Chandrasekhar,J.;Madura,J.D.;Impey,R. W.;K lein,M.L.J.Chem.Phys.1983,79(2),926.doi:10.1063/ 1.445869
(26)Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98(12), 10089.doi:10.1063/1.464397
(27)Berendsen,H.J.C.;Postma,J.P.M.;Vangunsteren,W.F.; Dinola,A.;Haak,J.R.J.Chem.Phys.1984,81(8),3684.doi: 10.1063/1.448118
(28)Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graph.Model. 1996,14(1),33.doi:10.1016/0263-7855(96)00018-5
(29)Delano,W.L.The PyMOLMolecularGraphicsSystem;Delano Scientific:San Carlos,CA,2002.
(30)Mhlongo,N.N.;Skelton,A.A.;Kruger,G.;Soliman,M.E.S.; Williams,I.H.Proteins2014,82(9),1747.doi:10.1002/ prot.24528
(31)Mohamed,I.P.K.;Subramani,K.Acta Phys.-Chim.Sin.2009, 25,2357.[Mohamed,I.P.K.,Subramani,K.物理化学学报, 2009,25,2357.]doi:10.3866/PKU.WHXB20091131
(32)Huang,Q.;Opitz,R.;Knapp,E.W.;Herrmann,A.Biophys.J. 2002,82(2),1050.
(33)Sondergaard,C.R.;Olsson,M.H.M.;Rostkowski,M.;Jensen, J.H.J.Chem.Theory Comput.2011,7(7),2284.doi:10.1021/ ct200133y
(34)Seeber,M.;Cecchini,M.;Rao,F.;Settanni,G.;Caflisch,A. Bioinformatics2007,23(19),2625.doi:10.1093/bioinformatics/ btm378
(35)Saha,B.C.J.Ind.M icrobiol.Biot.2001,27(4),241.doi: 10.1038/sj.jim.7000184
(36)Chavez,R.;Bull,P.;Eyzaguirre,J.J.Biotechnol.2006,123(4), 413.doi:10.1016/j.jbiotec.2005.12.036
(37)Eswar,N.;Eram ian,D.;Webb,B.;Shen,M.Y.;Sali,A.Protein StructureModelingwith MODELLER;Humana Press:New York,2008.
(38)Kirschner,K.N.;Yongye,A.B.;Tschampel,S.M.;Gonzalez-Outeirino,J.;Daniels,C.R.;Foley,B.L.;Woods,R.J. J.Comput.Chem.2008,29(4),622.doi:10.1002/jcc.20820
(39)Shallom,D.;Leon,M.;Bravman,T.;Ben-David,A.;Zaide,G.; Belakhov,V.;Shoham,G.;Schomburg,D.;Baasov,T.;Shoham, Y.Biochemistry 2005,44(1),387.doi:10.1021/bi048059w
(40)Yu,H.B.;Griffiths,T.M.Phys.Chem.Chem.Phys.2014,16 (12),5785.doi:10.1039/c4cp00351a
Mo lecu lar Dynam ics Sim u lation o f Ac tive-Sites o f Retaining and Invertingβ-Xy losidases
ZHANG Jun-Wei1ZHOU Jun-Gang1,*LÜHong1,2HUANG Qiang1,2,*
(1State Key Laboratory ofGenetic Engineering,SchoolofLife Sciences,ShanghaiEngineering Research Center of Industrial M icroorganisms,Fudan University,Shanghai200433,P.R.China;2ShanghaiCollaborative Innovation Centerfor Biomanufacturing Technology,Shanghai200237,P.R.China)
Xylans are im portantas potential renewable energy sources.In recentyears,there has therefore been interestin im proving theirdegradation efficiencies.β-Xylosidases are key enzymes for xylan degradation; these enzymes are classified,based on their hydrolysis mechanisms,as retaining or inverting enzymes. Althoughmuch research has been devoted to understanding retaining and invertingmechanisms,little is known about their differences in solution.We used molecular dynam ics(MD)simulations w ith explicit solvent representation to study the dynam ic behaviors of the active-sites of four typicalβ-xylosidases by analyzing the distances between two catalytic am ino acids and the p Kavalues ofproton-donoram ino acids.The results show that the distance between the catalytic am ino acidswith inverting enzymes is about0.8-1.0 nm,which is greater than that for retaining enzymes,i.e.,0.5-0.6 nm.This is consistentw ith previous results based on the crystal structures ofglycosidases.We found that the p Kaof the retaining proton donors aremodulated by interactions w ith neighboring am ino acids,enabling sw itching between low and high values.Such a p Kasw itch is neededfor the double-disp lacementmechanism of retaining enzymes.In contrast,inverting proton donors,modulated by interactions w ith neighboring glutam ic acids,have only high p Kavalues.Thismay be im portant in proton capture from the solventby donors,andmay facilitate the single-displacementmechanism of inverting enzymes. This study provides new insights into the hydrolysismechanisms ofβ-xylosidases,and w illtherefore be useful in improving the efficiency and app lications ofβ-xylosidases.
β-Xylosidase;Catalyticmechanism;Molecularmodelling;Proton donor;p Kavalue
O643;Q71
icle]
10.3866/PKU.WHXB201504151 www.whxb.pku.edu.cn
Received:February 16,2015;Revised:April13,2015;Published onWeb:April15,2015.
∗Corresponding authors.HUANG Qiang,Email:huangqiang@fudan.edu.cn;Tel:+86-21-51630589.ZHOU Jun-Gang,Email:zhoujg@fudan.edu.cn; Tel:+86-21-51630578.
The projectwassupported by the ShanghaiNaturalScience Foundation,China(13ZR1402400)and NationalNatural Science Foundation of China (31200022).
上海市自然科学基金(13ZR1402400)及国家自然科学基金(31200022)资助项目
©Editorialofficeof Acta Physico-Chim ica Sinica
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
国际放射医学核医学杂志(2020年4期)2020-07-27 01:53:28
高中数理化(2016年19期)2016-11-14 08:02:48
当代化工研究(2016年7期)2016-03-20 16:21:50
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
实用器官移植电子杂志(2015年1期)2015-04-02 14:58:26
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
