
低磷酸酶血症的研究进展
2015-12-26 00:21:32许莉军姜艳夏维波
国际内分泌代谢杂志 2015年3期
许莉军 姜艳 夏维波
低磷酸酶血症的研究进展
许莉军 姜艳 夏维波
低磷酸酶血症(HPP)是一种罕见的以骨和(或)牙齿矿化障碍,伴有血清碱性磷酸酶活性降低为特征的遗传性疾病。该病临床异质性强,容易造成漏诊和误诊。诊断主要依赖于临床表现、血清碱性磷酸酶降低及影像学特征。ALPL基因突变是诊断低磷酸酶血症必不可少的条件。HPP患者不建议使用维生素D和双膦酸盐。酶替代疗法将是未来几年最有前景的治疗方法。
低磷酸酶血症;ALPL基因;治疗
低磷酸酶血症是一种罕见的以骨和(或)牙齿矿化障碍,伴有血清碱性磷酸酶活性降低为特征的遗传性疾病[1]。该病于1935年首次由Chown描述,并于1948年由加拿大儿科医生Rathbun命名,故又称Rathbun综合征[2]。据估计,严重型低磷酸酶血症欧洲发病率为 1:300 000,加拿大为 1:100 000[1,3]。由于低磷酸酶血症临床异质性强,容易造成漏诊和误诊。本文旨在对低磷酸酶血症的研究进展进行综述。
1 发病机制
低磷酸酶血症是由编码组织非特异性碱性磷酸酶(TNSALP)的 ALPL(the liver/bone/kidney alkaline phosphatase gene)基因突变所引起的一种遗传性骨病。ALPL基因位于1p36.1-p34,包含12个外显子,编码长度超过50 kb[1]。该基因编码位于细胞膜上的一种磷酸单酯酶TNSALP,后者由524个氨基酸组成,包含活性区、N端螺旋区、同型二聚体结合区、冠状区及钙结合位点5个结构域[4]。TNSALP广泛存在于各种组织中,尤以肝脏、骨骼和肾脏分布较多,以同型二聚体形式存在时具有活性。正常情况下,TNSALP通过共翻译转运进入内质网后,经正确的折叠和组装后形成具有活性的同型二聚体,TNSALP与N端连接寡糖,通过C端的糖基磷脂酰肌醇锚定在内质网内侧,TNSALP从内质网转运到细胞表面的过程中,与其连接的寡糖经过加工后可抵抗内源性β-N-乙酰葡糖胺糖苷酶的分解,最终通过糖基磷脂酰肌醇锚定在细胞膜上。任何引起蛋白质折叠、组装、调节或转运异常的ALPL基因突变,均可引起基质小泡上TNSALP数量和性质的改变,从而引起低磷酸酶血症[5]。
TNSALP参与了焦磷酸盐的水解,但其对骨骼矿化的确切作用尚不清楚[6]。目前认为,焦磷酸盐在TNSALP作用下生成的无机磷与钙结合形成羟基磷灰石,具有促进骨骼矿化的作用;当血清碱性磷酸酶活性下降,不足以释放足够的磷酸与钙结合形成羟基磷灰石,大量血钙无法以磷酸钙的形式在骨内沉积,最终导致血钙升高。同时造成TNSALP的底物如无机焦磷酸盐、吡哆醛-5-磷酸及磷酸氨基乙醇的堆积。焦磷酸盐是骨骼矿化抑制剂,过多的焦磷酸盐抑制骨骼矿化,从而引起骨骼矿化障碍。
2 基因型与表型的关系
ALPL基因是低磷酸酶血症的唯一致病基因,95%的低磷酸酶血症患者均可检测到ALPL基因突变,未检测到的ALPL基因突变可能位于内含子区或调节区,也可能存在新的致病基因[7]。低磷酸酶血症的突变类型主要为错义突变,尚未发现热点突变。迄今为止,已报道ALPL基因突变300种,大部分患者来自欧洲、北美、澳大利亚和日本人群(http://www.sesep.uvsq.fr/03_hypo_mutations.php),仅有 1 例低磷酸酶血症患者为非洲黑人血统[8]。
许多研究发现,低磷酸酶血症的严重程度与TNSALP活性及碱性磷酸酶水平具有良好的相关性[4,9]。严重型低磷酸酶血症患者的ALPL基因突变发生在维持TNSALP活性的关键位点上,影响重要区域的氨基酸残基,使TNSALP不能进入细胞膜,从而在高尔基体聚集,导致TNSALP活性丧失;而轻型低磷酸酶血症的ALPL基因突变未发生在维持TNSALP活性的关键位点上,对TNSALP活性影响较小,TNSALP可部分地到达细胞膜发挥作用[10-11]。
严重型低磷酸酶血症患者以常染色体隐性方式遗传,而轻型以常染色体显性或隐性方式遗传。研究发现,临床表现越轻,以染色体显性方式遗传的可能性越大[12]。TNSALP以同型二聚体结构存在时具有活性,当同型的单体发生突变形成异二聚体时,TNSALP活性下降,推测在常染色体显性遗传性HPP中显性负效应发挥了重要作用[13]。Lia-Baldini等[14]发现,CFP/YFP标记的TNSALP质粒转染到大肠杆菌细胞,通过共聚焦荧光显微镜分析发现突变体G232V蛋白使野生型蛋白不能进入细胞膜发挥作用,认为显性负效应在低磷酸酶血症显性遗传中发挥重要作用。
部分杂合突变的低磷酸酶血症患者具有典型的临床表现,而其父母没有类似症状,提示该病可能有外显率不全或自发缓解趋势[3]。Taketani等[15]研究发现,相同的基因型由于发病年龄不同而表现为不同的临床类型。同一低磷酸酶血症家系内存在基因型相同而临床表型不同的现象,考虑可能和环境因素、表观遗传学或调节基因有关[7]。
3 临床分型及各型临床表现
3.1 新生儿致死型 该型是低磷酸酶血症最严重的类型。胎儿期因严重的骨矿化障碍常出现死胎。一些婴儿因胸部畸形导致肺发育不良,常于出生数天因呼吸系统并发症而死亡。有的患儿表现为窒息、抽搐发作及长骨短。
3.2 围产期轻型 围产期轻型表现为四肢短小、弯曲,骨骼畸形及骨矿化障碍可自发缓解,预后良好。
3.3 婴儿型 婴儿型是低磷酸酶血症严重类型之一。出生时可表现正常,在出生6个月内出现临床症状。由于胸部佝偻病样畸形,常出现呼吸系统并发症。部分患者有高钙血症的表现如烦渴、多尿、喂养困难、便秘、恶心。该型常有颅缝早闭,从而导致颅内高压。
3.4 儿童型 出生6个月后发病,临床表现多样,可表现为佝偻病、乳牙脱落早(常于5岁前脱落)、身材矮小、走路延迟及鸭步态等。除颅缝早闭外,其他临床表现有自限性趋势,但常于成年后再发。Taketani等[15]报道,日本低磷酸酶血症患儿中智力障碍、耳聋、身材矮小发生比例明显高于西方人群。
3.5 成年型 中年起病,表现为负重部位的骨痛、应力性骨折及恒牙早发脱落等。该型也可表现为软骨钙质沉着病、骨关节病[16]。过量的焦磷酸盐沉积于韧带、关节周围可引起二水焦磷酸钙结晶沉积症、焦磷酸盐关节病变、假性痛风发作和钙化性关节周围炎,常见累及部位为肩、髋、肘、膝盖及手腕等[17]。
3.6 牙型 表现为牙齿过早脱落、牙槽骨量减少、牙髓腔变大,伴(不伴)龋齿,无骨骼异常表现。
4 诊断
目前没有低磷酸酶血症的统一诊断标准。主要依赖临床表现、实验室检查及影像学特征进行诊断。ALPL基因突变是诊断低磷酸酶血症必不可少的条件[4]。
除了假性低磷酸酶血症外,所有类型的低磷酸酶血症均表现为血清碱性磷酸酶活性降低。血清碱性磷酸酶活性降低需除外早期妊娠、双膦酸盐或其他强有力的抗骨吸收药物的使用、甲状腺功能减退及贫血。血清吡哆醛-5-磷酸及尿中磷酸氨基乙醇浓度升高有助于该病的诊断。
年龄和临床分型不同,低磷酸酶血症患者影像学表现不同。新生儿致死型X线表现为严重骨矿化不足(图1.A),前臂或股骨突出皮肤的软骨样骨刺,对该病具有诊断意义;婴儿型和儿童型可表现为佝偻病改变如干骺端杯口样改变、下肢弯曲、干骺端增宽凹陷(图1.B)、放射线透亮区(图1.C)及临时钙化带变薄或消失;婴儿型可有颅缝早闭、头颅狭小和颅压升高的X线征象;成年型可见假骨折线(图1.D)、软骨钙质沉着症(图1.E)。
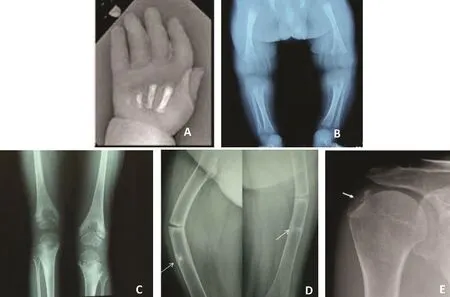
图1 不同临床分型低磷酸酶血症的影像学表现
5 治疗及预后
目前尚无低磷酸酶血症的根治方法。既往研究认为,饮食中限制磷的摄入对该病是有效的[4]。有研究发现,非甾体抗炎药可改善儿童型低磷酸酶血症患者的临床表现,尤其是缓解骨痛,同时可减轻由于该病引起的炎性反应[18]。Doshi等[19]对1例出现双侧股骨应力性骨折的成人低磷酸酶血症患者使用特立帕肽(甲状旁腺激素 1-34,20 μg/d),治疗8周后患者骨痛明显缓解,16个月双侧股骨骨折愈合,生化检查提示尿中Ⅰ型胶原交联氨基末端肽升高11%,而血中骨源性碱性磷酸酶水平并未升高。Schalin-Jäntti等[20]对 2例成年低磷酸酶血症患者分别给予重组人甲状旁腺激素(甲状旁腺激素1-84,100 μg/d)治疗 7 个月和 18 个月,血碱性磷酸酶水平分别升高6.8倍和2.7倍,2例患者骨痛缓解,骨折愈合。这些结果提示,重组人甲状旁腺激素可改善低磷酸酶血症患者的临床症状,但长期重组人甲状旁腺激素治疗的安全性尚需进一步观察。由于三磷酸核苷在细胞膜糖蛋白-1(PC-1)的作用下生成焦磷酸盐,PC-1抑制剂可抑制焦磷酸盐的产生,从而促进骨骼矿化。Hessle等[6]研究发现,ALPL基因敲除小鼠当出现PC-1基因失活突变时骨骼矿化不良得以纠正,小鼠恢复正常的表型,提示PC-1抑制剂或许是治疗低磷酸酶血症的有效措施。有报道称,骨髓细胞和间充质干细胞移植可改善低磷酸酶血症患者的临床症状,但长期的有效性和安全性尚需进一步评估[21-22]。Yadav 等[23]报道,ALPL 基因敲除小鼠出现牙釉质发育不良,临床表现类似婴儿型低磷酸酶血症,给予重组人TNSALP治疗后牙釉质发育不良明显改善。ENB-0040是一种靶向作用于骨骼的重组人TNSALP,在一项入组了11例严重型低磷酸酶血症患儿的多种族、开放的研究中,ENB-0040治疗1年后11例低磷酸酶血症患儿佝偻病得以治愈,肺功能明显改善,血焦磷酸盐和吡哆醛-5-磷酸水平降至正常,无低血钙及异位钙化等不良反应[24]。Kosnik-Infinger等[25]对4例合并颅缝早闭的低磷酸酶血症患儿给予ENB-0040及手术干预治疗,患儿骨骼矿化不良及神经系统症状明显改善。目前该药物正在进行青少年及成人低磷酸酶血症患者的Ⅱ期临床试验。酶替代疗法将是未来几年最有前景的治疗方法[23,26]。
由于低磷酸酶血症患者血钙和血磷水平升高或正常,25-羟维生素D3正常,因此维生素D治疗常无效,甚至可出现临床症状的加重[27]。低磷酸酶血症患者禁用双膦酸盐,后者可加重骨骼矿化障碍[28]。因此骨质疏松的患者使用双膦酸盐前应首先排除低磷酸酶血症的可能。
该病预后与发病年龄和疾病的严重程度有关。发病年龄越早,预后越差[29]。新生儿恶化型低磷酸酶血症因骨骼严重矿化不足常在数天或数周内死亡,一半的婴儿型低磷酸酶血症死于呼吸道并发症[1]。成年型及牙型低磷酸酶血症患者寿命正常,预后良好。
总之,低磷酸酶血症临床表现异质性强,给诊断带来很大困难。对于临床上疑诊为低磷酸酶血症的患者,需检测血清碱性磷酸酶水平。若血清碱性磷酸酶水平下降,需进一步行ALPL基因筛查。对低磷酸酶血症患者进行分子遗传学检测不仅有利于早期诊断,而且对该家庭进行遗传咨询和产前诊断提供了理论依据。关于低磷酸酶血症,在发病机制及治疗方面已经取得很大进步,但仍有很多问题尚待解决。进一步的研究应集中于TNSALP影响骨骼矿化的机制及轻型低磷酸酶血症的患病率,同时应对重组人TNSALP替代治疗进行大样本量、多中心、随机、对照研究以评估该药的安全性、有效性。
[1]Mornet E.Hypophosphatasia [J].Orphanet J Rare Dis,2007,2:40.
[2]RATHBUN JC.Hypophosphatasia:a new developmental anomaly[J].Am J Dis Child,1948,75(6):822-831.
[3]Mornet E,Yvard A,Taillandier A,et al.A molecular-based estimation of the prevalence of hypophosphatasia in the European population[J].Ann Hum Genet,2011,75(3):439-445.
[4]Mornet E.Hypophosphatasia[J].Best Pract Res Clin Rheumato,2008,22(1):113-127.
[5]Sultana S,Al-Shawafi HA,Makita S,et al.An asparagine at position 417 of tissue-nonspecific alkaline phosphatase is essential for its structure and function as revealed by analysis of the N417S mutation associated with severe hypophosphatasia[J].Mol Genet Metab,2013,109(3):282-288.
[6]Hessle L,Johnson KA,Anderson HC,et al.Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization[J].Proc Natl Acad Sci U S A,2002,99(14):9445-9449.
[7]Mornet E.Genetics of hypophosphatasia[J].Clinic Rev Bone Miner Metab,2013,11:71-77.
[8]Whyte MP,Essmyer K,Geimer M,et al.Homozygosity for TNSALP mutation 1348c>T(Arg433Cys)causes infantile hypophosphatasia manifesting transient disease correction and variably lethal outcome in a kindred of black ancestry[J].J Pediatr,2006,148(6):753-758.
[9]Zurutuza L,Muller F,Gibrat JF,et al.Correlations of genotype and phenotype in hypophosphatasia[J].Hum Mol Genet,1999,8(6):1039-1046.
[10]Satou Y,Al-Shawafi HA,Sultana S,et al.Disulfide bonds are critical for tissue-nonspecific alkaline phosphatase function revealed by analysis of mutant proteins bearing a C(201)-Y or C(489)-S substitution associated with severe hypophosphatasia[J].Biochim Biophys Acta,2012,1822(4):581-588.
[11]Mornet E,Stura E,Lia-Baldini AS,et al.Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization[J].J Biol Chem,2001,276(33):31171-31178.
[12]Fauvert D,Brun-Heath I,Lia-Baldini AS,et al.Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles[J].BMC Med Genet,2009,10:51.
[13]Lia-Baldini AS,Muller F,Taillandier A,et al.A molecular approach to dominance in hypophosphatasia[J].Hum Genet,2001,109(1):99-108.
[14]Lia-Baldini AS,Brun-Heath I,Carrion C,et al.A new mechanism of dominance in hypophosphatasia:the mutated protein can disturb the cell localization of the wild-type protein[J].Hum Genet,2008,123(4):429-432.
[15]Taketani T,Onigata K,Kobayashi H,et al.Clinical and genetic aspects of hypophosphatasia in Japanese patients[J].Arch Dis Child,2014,99(3):211-215.
[16]Berkseth KE,Tebben PJ,Drake MT,et al.Clinical spectrum of hypophosphatasia diagnosed in adults[J].Bone,2013,54(1):21-27.
[17]Guanabeñs N,Mumm S,Möller I,et al.Calcific periarthritis as the only clinical manifestation of hypophosphatasia in middleaged sisters[J].J Bone Miner Res,2014,29(4):929-934.
[18]Girschick HJ,Schneider P,Haubitz I,et al.Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia[J].Orphanet J Rare Dis,2006,1:24.
[19]Doshi KB,Hamrahian AH,Licata AA.Teriparatide treatment in adult hypophosphatasia in a patient exposed to bisphosphonate:a case report[J].Clin Cases Miner Bone Metab,2009,6(3):266-269.
[20]Schalin-Jäntti C,Mornet E,Lamminen A,et al.Parathyroid hormone treatment improves pain and fracture healing in adult hypophosphatasia[J].J Clin Endocrinol Metab,2010,95(12):5174-5179.
[21]Taketani T,Kanai R,Abe M,et al.Therapy-related Ph+leukemia after both bone marrow and mesenchymal stem cell transplantation for hypophosphatasia[J].Pediatr Int,2013,55(3):e52-e55.
[22]Whyte MP,Kurtzberg J,McAlister WH,et al.Marrow cell transplantation for infantile hypophosphatasia[J].J Bone Miner Res,2003,18(4):624-636.
[23]Yadav MC,de Oliveira RC,Foster BL,et al.Enzyme replacement prevents enamel defects in hypophosphatasia mice[J].J Bone Miner Res,2012,27(8):1722-1734.
[24]Whyte MP,Greenberg CR,Salman NJ,et al.Enzyme-replacement therapy in life-threatening hypophosphatasia[J].N Engl J Med,2012,366(10):904-913.
[25]Kosnik-Infinger L,Gendron C,Gordon CB,et al.Enzyme replacement therapy for congenital hypophosphatasia allows for surgical treatment of related complex craniosynostosis:a case series[J].Neurosurg Focus,2015,38(5):E10.
[26]Oikawa H,Tomatsu S,Haupt B,et al.Enzyme replacement therapy on hypophosphatasia mouse model[J].J Inherit Metab Dis,2014,37(2):309-317.
[27]Mohn A,De Leonibus C,de Giorgis T,et al.Hypophosphatasia in a child with widened anterior fontanelle:lessons learned from late diagnosis and incorrect treatment[J].Acta Paediatr,2011,100(7):e43-e46.
[28]Sutton RA,Mumm S,Coburn SP,et al."Atypical femoral fractures"during bisphosphonate exposure in adult hypophosphatasia[J].J Bone Miner Res,2012,27(5):987-994.
[29]Faruqi T,Dhawan N,Bahl J,et al.Molecular,phenotypic aspects and therapeutic horizons of rare genetic bone disorders[J].Biomed Res Int,2014,2014:670842.
Research progress of hypophosphatasia
Xu Lijun,Jiang Yan,Xia Weibo.Department of Endocrinology,Key Laboratory of Endocrinology,Ministry of Health,Beijing Union Medical College Hospital,Chinese Academy of Medical Sciences Peking Union Medical College,Beijing 100730,China
Xia Weibo,Email:xiaweibo@medmail.com.cn
Hypophosphatasia is a rare inherited disorder characterized bydefective bone and/or dental mineralization,and decreased serum alkaline phosphatase activity.The disease can be easily missed diagnose and misdiagnosed.The diagnosis of hypophosphatasia is based on clinical manifestations,decreased alkaline phosphatase level and radiological examinations.Screening for mutations in the ALPL gene is essential to diagnose the disease.Vitamin D and bisphosphonates are not suggested to use.Enzyme replacement therapy will be certainlythe most promisingchallenge ofthe next fewyears.
Hypophosphatasia;ALPLgene;Therapy
(Int J Endocrinol Metab,2015,35:211-214)
10.3760/cma.j.issn.1673-4157.2015.03.020
国家自然科学基金资助项目(81070687,81170805)
100730 北京,中国医学科学院,北京协和医学院,北京协和医院内分泌科,卫生部内分泌重点实验室
夏维波,Email:xiaweibo@medmail.com.cn
2014-12-04)
猜你喜欢
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:45
电镀与环保(2016年2期)2017-01-20 08:15:25
浙江农业科学(2016年11期)2016-05-04 04:16:45
广西林业科学(2016年1期)2016-03-20 05:33:00
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
电源技术(2015年2期)2015-08-22 11:27:56
医学研究杂志(2015年8期)2015-06-22 14:00:57
无机化学学报(2014年12期)2014-02-28 17:33:54
中国医学科学院学报(2013年6期)2013-03-11 20:26:04
