
橡胶试验方法(五十二)
——摘自日本《ゴム試驗法》
2015-12-24 06:42:49王作龄张卓娅编译
橡塑资源利用 2015年2期
王作龄,张卓娅 编译
橡胶试验方法(五十二)
——摘自日本《ゴム試驗法》
王作龄,张卓娅 编译
5.2 试样制作的一般顺序
5.2.1 原料胶乳取样和处理方法
关于胶乳试验用试样,如上所述作为JIS K6387—1(胶乳一第1部分:取样),对应ISO 123中IDT的标准进行制定。
在橡胶与水的密度差大时,胶乳静置后通过膏化或沉降,存在上下产生浓度差的倾向。因此取样前必须充分搅拌,制备有再现性的代表试样。对此的取样方法在标准中详述。
按每种容器规定的方法。对整体胶乳进行搅拌,从胶乳容器自上而下各位置取样。从各位置取的胶乳总固形分在0.5%以内一致时被视为均匀,然后将其合为一体进行充分搅拌混合。之后置于密封容器中保存用于试验。容器的材质应使用对胶乳无影响的材料。另外,对于搅拌胶乳的装置也要注意不能含铜。
取样时,必须确认并记录胶乳中杂质或凝块含量等情况。此外,对于测定凝块含量以外使用的情况,测定前必须充分搅拌,用(180±10)μm的不锈钢网或使用对胶乳呈惰性的合成布进行过滤之后再使用。
5.2.2 干燥与硫化胶膜处理方法
在评价原料胶乳或配合胶乳,或者在评价胶乳配合剂的效果时,制备干燥腔膜,或者在一定温度下变更硫化时间对干燥胶膜进行硫化。对制作得到的硫化胶膜进行拉伸试验、撕裂试验,溶胀试验、永久变形试验、老化试验和浸渍试验等,进行对比研究。
5.2.2.1 原料胶乳干燥胶膜制作方法
关于由原料胶乳制作干燥胶膜的方法,在ISO 498中进行了规定。
在ISO 498中,使用100~150mm2的带框玻璃板(框尺寸为宽6mm、厚度1.0mm)和水平板制作干燥胶膜。关于试样制备,先将比计算量稍多的胶乳进行过滤、除泡,再将这种胶乳注入胶模制作模具,静置1min后,为了标准或呈水平,用棒以25mm/s以下的速率刮模具表面,以除去多余的胶乳。流延的胶膜在无粉尘的通常的大气中进行干燥。于室温下干燥后,在35℃以下的干燥器中完成干燥,然后尽量不接触手而将试样从玻璃板上剥下。然后,将胶膜翻过来平放在玻璃纸上,于35℃的地方至少再放置24h,当这一面完全干了时用相同薄片覆盖,放进干燥器或密封的容器中防止吸湿,试验之前保存在阴冷处。
胶乳总固形物含量不足62%时直接使用,若在62%以上则用蒸馏水稀释至61.5%,然后用(0.185±0.01)mm的尼龙网或不锈钢网过滤,静置5min以上,用滤纸除掉其表皮或气泡等。所得干燥胶膜的厚度,若胶乳浓度是62%则大致为1mm。
5.2.2.2 配合胶乳干燥胶膜制备方法
配合胶乳的干燥胶膜和硫化胶膜的标准制备方法只有日本橡胶协会标准学会标准(SRIS 3201)中进行了规定。现在该标准正在废除,而在此以SRIS 3201为基础对胶乳的干燥胶膜制作方法进行说明。用于评价干燥胶膜或硫化胶膜的物理试验,没有专门针对胶乳薄膜的,可根据JISK 6251(硫化橡胶和热塑性橡胶—拉伸性能的求法)进行。
对于用胶乳制造与干胶试样相同厚度的试样胶膜存在各种各样的问题。特别对于制作配合胶乳的胶膜,与制作原料胶乳的胶膜相比,存在着配合剂沉积、分离引起不均匀化问题。要制作和干胶相同厚度的胶膜时,因干燥需要很长时间,所以胶乳中的配合剂会产生沉积,或干燥中进行硫化,或产生龟裂等问题。因此,为了缩短干燥时间,将胶膜厚度规定为0.5~1.0mm较适宜,在SRIS 3201中以该厚度范围为标准规定了干燥胶膜的标准制备方法。该厚度范围不仅是从胶膜制备方面考虑,也是以物理试验数据的再现性方面考虑的。但对于黏度低、湿凝胶强度显著低的或收缩率大的合成胶乳,即使以该范围的厚度也会发生配合剂沉积或龟裂面得不到良好的胶膜。
为防止配合剂沉积面使用增黏剂时会引起胶乳的分离(由膏化引起)。配合组成因膏化进行变化,使强度等性能受到影响,因此解析结果变得复杂。这种情况,如果实际加工以凝固形式进行时,由凝固法制作胶膜就比较方便。因为那样的目的,SRIS 3201中在规定流延法的同时也规定了由凝固法制作胶膜的方法,但对于没有那种加工工序的情况,由凝固法制模由于对膜结构、凝固剂的硫化性和老化性影响的问题一般不理想。
对于那种情况,不得不使流延胶膜更薄。胶膜变薄时(如0.1~0.2mm)干燥时间显著缩短。其结果,沉积问题也的确变得不显著,产生的龟裂因模型的支承力也极大减少。但是,模具工作面的伤痕、胶乳中的杂质或微小气泡、配合剂聚集块等的缺陷部分、试样冲模的伤痕或拉伸试验机固定试样装置等这些因素的影响增大,测定数据的波动存在增大倾向,因此要注意。
也有在使用石膏等多孔物质的模上进行流动的提案,但使用这种模具时,水和水溶性物质一起被除去,存在组成发生变化的缺点。另外数次使用后,模具表面变粗糙,或者部分附着水锈,不能实现表面状态的再现性。
流延胶膜的干燥,当大气湿度低时表面干燥显著。表面产生干燥胶膜时,因该胶膜难以透过水分则干燥逐渐减慢,所以最初提供60%~70%程度的湿度,达到缩短整个干燥时间的效果。
在板上,特别对于冬季室温低的场合下的干燥有时要操心。因此在考虑使用利用低温面热体的胶膜处理装置。干燥的胶膜多数对湿气非常敏感,因此不能适宜保存状态时干燥胶膜的物性就有可能发生显著变化。对此必须注意。
5.2.2.3 干燥胶膜的硫化
干燥胶膜从模具上剥下时在自由表面一侧做上标志,然后在表面上压合具有硫化温度以上耐热性而且不含向橡胶中迁移性物质的塑料薄膜等。在对该干燥胶膜硫化时,将保护薄膜悬挂在预先调整到规定硫化温度的强制循环式干热型恒温槽中,然后合上门进行硫化。该操作尽量迅速进行,必须注意避免温度显著降低。若可能的话,硫化装置最好是使用关门时具有动作的辅助加热装置的快建返回式。此外对于胶膜悬挂,至少离周围壁画5cm以上,而且互相保持适当的距离,必须考虑硫化装置内的温度分布和保证通风状态不能出现显著不稳定。
此外,使用保护膜的目的是防止附着杂质和硫化中表面氧化。但是,干燥胶膜中残留水分时,对于不透薄膜的情况,在薄膜与橡胶试样胶膜之间存积水蒸气,损坏硫化橡胶试样表面的光滑性,因此必须注意。这种情况特别对于皂含量较高而难以干燥并且硫化温度高的SR胶乳需要特别注意。硫化结束的试样置于干燥器中,保存在阴冷处供试验使用。
5.3 原料和配合胶乳的试验方法
5.3.1 原料胶乳的通用试验方法
5.3.1.1 总固形物测定
原料胶乳总固形物的测定方法是由百分率表示称量的胶乳在规定干燥温度下的不挥发分含量,以ISO 124实行标准化。作为JIS标准的JIS K6387—2(橡胶胶乳一第2部;总固形物含量的求法)以ISO 124的IDT标准发行。
胶乳的加热方法分为大气压下加热方法和减压下加热方法两种。NR胶乳只适用大气压下加热方法,SR胶乳可适用大气压下和减压下两种加热方法。
对于大气压下的测定,用平皿容器称取试样胶乳约(2.0±0.5)g(准确至0.1mg),然后将胶乳均匀分布于整个平皿容器。此时,根据需要加入蒸馏水1cm3稀释胶乳。将平皿容器中的胶乳以(70±2)℃干燥至16h或以(105±5) ℃干燥2h,在干燥箱内冷却后测定重量。测定后再次以(70±2)℃干燥30min和以(105±5)℃干燥15min,然后测定重量。进行反复干燥、测定后,将连续两次测定值之差小于0.5mg的测定值作为结果。
对于减压测定,在平皿容器内称取试样胶乳(1.0±0.2)g,然后用1cm3蒸馏水进行稀释,使容器覆盖。在低于20kPa的压力和(125±2)℃的温度下对平皿容器中的胶乳干燥45~65min,在干燥箱内冷却后测定重量。反复进行干燥,测定,将两次测定值之差在0.5mg以下的测定值作为结果。在测定上,应将胶乳以同样厚度分布在容器的底面,为防止减压时起泡或飞散,逐渐减压很重要。总固形物测定成为交易或配方设计的基础,因此是重要的测定。但对于标准方法因费时间,所以考虑了种种快速测定方法,可用于实际的商业交易。那时,由于150℃以上高温干燥的分解等,而存在固形物变低的胶乳,因此要注意。
5.3.1.2 pH值测定
关于pH值测定,ISO 976中规定了玻璃电极pH计的测定方法。pH计的校正由pH7的标准液和pH4或pH9的标准液进行。测定温度规定为(23±3)℃,热带地区规定为(27±3)℃。测定方面重要的问题是,试验前将胶乳、pH标准液和洗涤用蒸馏水保存在试验室内,以便使温度一致。另外进行测定的胶乳必须充分搅拌均匀。
测定时先将由蒸馏水洗过的玻璃电极由少量的试样洗涤,而后充分浸入试样。测定值稳定时读取数值,然后用新试样再测定一次。测定差在0.1以下就将其平均值作为测定值。如果两次测定的差在0.1以上,就再测定两次。
5.3.1.3 黏度测定
胶乳黏度测度在ISO 1652中做了规定。关于胶乳黏度的测定,很早就使用各种装置测定。在黏度测定中,浓度高的胶乳由于表现非牛顿流动,所以存在测定值依测定方法而异的问题。另外即使同一黏度计,使用的锭子的形状尺寸和转速等不同时,测定值也不同。因此,在黏度测定结果中必须记录所用锭子的种类。在ISO 1652中规定了L型和R型两类黏度计,L型可用于2000mPa·s(2000cP)以下的测定,面R型可用于200mPa·s(200cP)以上的测定。
L型黏度计使用60r/min的转速,而R型黏度计使用20r/min的转速,采用20~30s后读数。图5-3-1是L型和R型黏度计的转子。
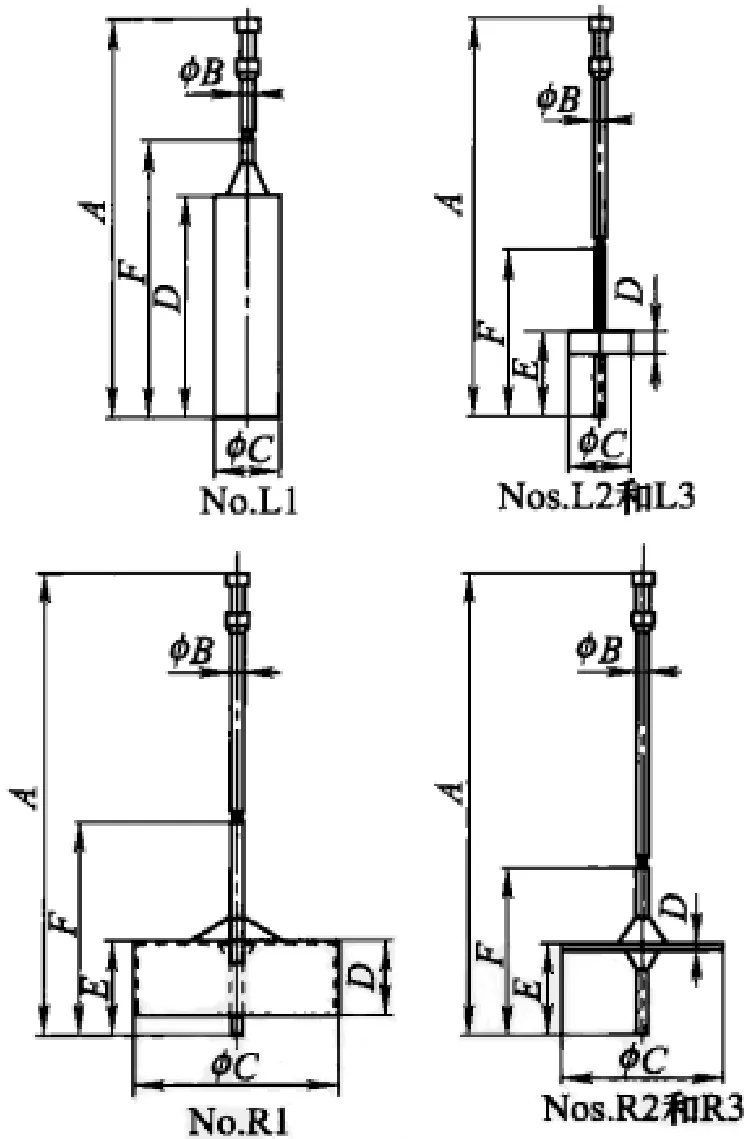
图5-3-1 黏度测定用转子(据lSO标准)
黏度测定中应注意的问题是,除去胶乳中的气泡,将锭子沉入胶乳中时不能附上气泡,温度恒定(ISO中规定为25℃和27℃)等。转速慢时由于固定指针的位置容易变化指针的度数,因此认为在指针通读取窗时压动控制杆的方法可以固定指针。黏度由读出的刻度数乘以由锭子种类决定的常数计算,单位用mpa·s(cP)裹示。
5.3.1.4 密度测定
胶乳密度测定在ISO 705中进行了规定。该标准是为了计算不能直接测定重量而已知容量NR胶乳的重量所规定的。而且,如果以计算已知容积胶乳的重量为目的,则对测定容积时混入的空气和含同量空气的试样进行测定很重要。因此,规定取样后应保持24h以上确认除去气泡。另外先将胶乳的温度调整到与测定容积时相同的温度后再进行测定。如果测定温度与测定容积时的温度不同时,则必须修正。
使用呈特殊形状的瓶进行测定。测定前先将胶乳静置24h以上除去气泡,再将该胶乳和蒸馏水的温度调整到容量测定时的温度。然后测定该胶乳和水一定体积的质量。将结果代入下式,算出胶乳在容积测定温度下的密度。如果不能调整温度时,则使用在试验室内适当的温度下的测定值和胶乳的膨胀系数进行修正。
胶乳密度按下式计算:
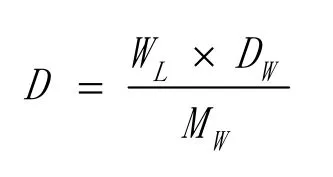
式中D—容积测定时温度下的胶乳密度,g/cm3;
WL—上述温度的比重瓶中胶乳的质量,g;
Mw—上述量度的蒸馏水的质量,g;
Dw—上述温度的水的密度,g/cm3。
5.3.1.5 凝块含量测定
凝块含量的测定方法以ISO 706进行标准化。
凝块含量测定按以下方法进行。对于NR胶乳的情况,将约200g胶乳由同量的5%油酸钾水溶液或5%月桂酸铵水溶液进行稀释,而对于SR的情况由5%的乙氧基化烷基苯酚水溶液进行稀释,然后测定重量(准确至1mg),并用湿的(180±10)μm的不锈钢网进行过滤。过滤后由稀释用的界面活性剂水溶液洗涤金属网,确认无胶乳。最后用蒸馏水充分洗涤,对残余物进行干燥(100~105℃),由对于总固形物含量的百分率表示。除了橡胶凝块外也含有胶乳薄膜或杂质等。
5.3.1.6 表面张力测定
胶乳表面张力的测定方法在ISO 1409中规定了铂环法的表面张力剥测定方法。测定值读取使用DuNouy型表面张力计,铂环使用规定尺寸铂—铱合金的。测定温度规定为(23±3)℃,热带地区规定为(27±3)℃.
测定中,调制胶乳很重要。对总固含量调整至40%或其以下,黏度特别高时调整至低于200mPa·s,然后进行测定。测定不在震动的地方,注意气泡和结皮,而且必须迅速进行。测定结果由下式计算,单位是N/m(dyne/em)。
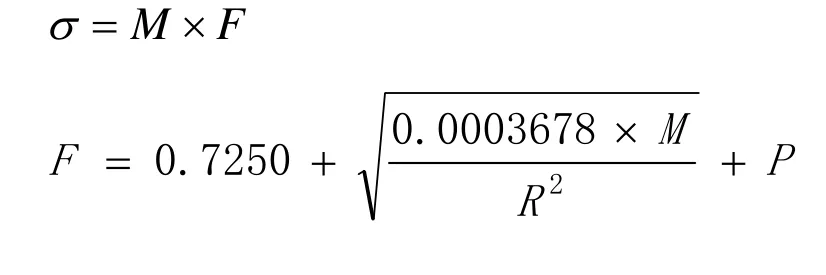
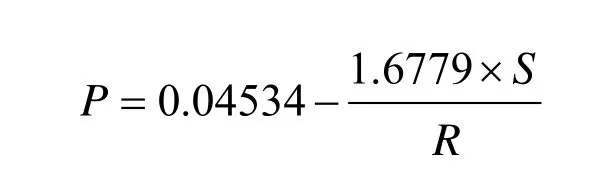
式中σ—表面张力;
M—表面张力计读数;
R—铂环半径,cm;
S—铂环丝半径,cm。
铂环法的表面张力测定法为了测定铂环离开液面时的力,对静态力进行测定。但考虑到加工等方面,需要评价动态过程的界面现象。作为动态表面张力的测定方法有液压法、悬滴法和液重法等。这些测定方法现在没有标准化。
5.3.1.7 电导率测定[2,3]
作为测定胶乳电导率的方法,没有特别规定标准化的试验方法。但是,用标准的物理化学方法可进行测定。胶乳电导率的测定特别是可用作表示NR胶乳的质量。这是因为非橡胶份水解产生的酸形成铵盐,NR胶乳变得不新鲜时电导率增加。因此认为,这和氢氧化钾值或挥发性脂肪酸含量有很大关系。
另外电导率测定可用于SR胶乳的皂含量测定或作为SR胶乳粒径测定方法之一的皂滴定法。
5.3.1.8 粒径和粒径分布测定
粒径或其分布的测定不仅是了解粒径分布的问题,而且也是关系黏度或浸透性等加工性能的重要性质。但是,作为其测定标准尚未规定,下述方法可作为粒径测定法使用。
(1)电子显微镜照相法[4,5]
和常用方法一样用显微镜进行检查照相,测定粒子的大小、形状和粒子分布。这种方法的优点不仅可以观察粒径的分布而且可以观察粒子的形状,但依聚合物的性质不同容易变形,因此存在测定值波动大的可能。为防止这种情况可进行溴化等。通过溴化虽可防止变形,但需要修正由溴化引起的膨胀。
(2)皂滴定法[6]
这是胶乳粒子表面在由皂的单子层完全覆盖前利用皂吸附的性质的方法,通过测定表面张力或电导率进行。因此,测定用皂的种类不明确时不适宜,而且由于以平均粒径得出结果,所以在粒径存在分布时有可能得不出有用的结果。
(3)光散射法[7,8]
胶乳粒子以在水介质中分散的状态进行布朗运动,而粒子的运动随着胶乳粒径增大而减小。利用这种性质对胶乳粒子进行激光照射,观测由胶乳粒子引起的散射光波动。用光子相关法解析这种波动,求出粒径或粒径分布。
除了这些方法外,还有离心沉淀法等。关于这些测定方法在此省略,但由这些方法得到的平均粒径值依测定方法而异。例如,电子显微镜法的平均粒径值是数量平均,皂滴定法的平均粒径值是面积平均,离子沉淀法的平均粒径值是重量平均。因此,对于粒径分布窄的胶乳,这些方法的测定值都一致,但对于粒径分布宽的放乳,这些方法的测定值之间产生相当大的差异。因此,也可将重均粒径与数均粒径之比规定为粒径分布的大致标准。
5.3.1.9 机械稳定度试验方法(MST)
机械稳定度测定有两种标准化方法。对于NR胶乳规定了ISO 35标准,对于SR胶乳规定了ISO 2006(高速测定)标准。另外对SR胶乳曾经以JIS规定了所谓马朗(Maron)式的低速测定标准。关于这些标准,在NR胶乳和SR胶乳的各自试验方法中有详细介绍。
5.3.1.10 化学稳定度试验方法[9]
胶乳中加入某物质时,随着时间产生凝块,有时整体进行胶凝化。胶乳的这种性质被称为化学稳定性。化学稳定性是确保胶乳加工时的稳定性和控制加工时引起胶凝化的指标,所以是重要的试验项目。因此,按胶乳的用途对种种物质的稳定性进行了试验。例如,对硅氟化钠、氯化钾、乙醇、甲苯及碳酸钙等液体和粉体等物质都试验了化学稳定性。但在一般配合剂中,ZnO(氧化锌)的不稳定作用最大,因此一般将添加氧化锌时黏度上升或出现凝固的时间定为化学稳定性尺度。化学稳定度试验方法分为ZOT试验,ZOV试验和ZST试验等。这些试验都是以NR胶乳为对象的试验。ZnO在氨和铵离子存在下形成络盐进行溶解,随着温度上升或pH值降低而放出Zn2+,从而使胶乳稳定化。SR胶乳多数不含氨,因此不生成锌络盐而不产生稳定化。但在与NR胶乳共混时是关系胶乳加工性的重要试验方法。下面简介化学稳定性试验的例子。
(1)ZOT试验(氧化锌增稠试验)[10]将总固形物含量规定为55%,使氨浓度精确降至0.05%,加入针对固形物含量为3%的ZnO的50%分散体。然后测定加ZnO 5min后和24h后胶乳的黏度,将其黏度上升分作为稳定性的指标。该试验由于pH值的微小差就能对试验结果影响很大,因此需要注意。该测定成为浸渍或挤出加工用胶乳的有益指标。
(2)ZOV试验(氧化锌黏度试验)[11]用500cm3烧杯取试样胶乳320g,其中加人20%油酸钾溶液10g,继之加入水9g,充分搅拌混合后置于恒温箱,将温度调至25℃。确认胶乳的黏度无变化后,添加l0%硫酸铵溶液10g和40% ZnO分散体25g,然后立即按秒表搅拌1min。添加ZnO后用B型黏度计(No.2转子,60rpm)进行测定。通常采用5min后的值作为ZOV值。ZOV的结果常用作Dunlop法泡沫橡胶生产中加工性的指标。ZOV在500~700之间为加工性良好,在1000以上为过于不稳定,在300以下为过于稳定。此外ZOV试验也可用于试验原料胶乳的保存性。
(3)ZST试验(氧化锌稳定度试验)[12]
这是存在氧化锌条件的MST试验,除了纯粹的化学稳定性外还含有机械稳定性的因素。因此正确一点说最好是称为机械化学稳定性。该试验方法的要点是,对于橡胶含量添加1%油酸钾,加福尔马林将pH值变成9.75~9.80,再加入蒸馏水将固形物含量调整至55%。用烧杯取该胶乳182g置于恒温箱(30℃),盖上盖子放置10~15min。然后一边搅拌一边在10min内直接添加5s氧化锌粉末。从开始加氧化锌起正确搅拌15min。60min后缓慢搅拌过滤,取80g于30℃下进行MST试验。ZST在150s以内表示过分不稳定,在150~300s之间表示稳定性良好,在300~500s之间表示高稳定。在500s以上表示是一种适于ZnO存在下长期保持的胶乳。
除这种方法以外,还有加入硅氟化钠测定达到凝固的时间的方法,添加一定浓度氧化钾水溶液测定达到凝固添加量的方法,以及用不同浓度氯化钾水溶液根据进行凝固的浓度测定稳定性的方法。化学稳定性的评价要在考虑了加工方法的基础上进行。
5.3.1.11 热稳定性测定
NR胶乳只要不含ZnO,即使在高温下也比较稳定。但添加ZnO后即使时间长短依温度而异,但最终都会产生不稳定化。存在ZnO时的热稳定性和化学稳定性相同。对于化学稳定性是测定室温下经时的增黏,而对于热稳定性是测定硫化时的凝固时间或一定时间的最低胶凝化温度。加热时凝固时间的测定有氧化锌稳定度和修正醋酸试验等。另外作为一定时间内最低胶凝化温度有临界稳定度试验(CST)、氧化锌热敏性试验和氧化锌号数试验(Zinc Oxide Number Test)等。
对于SR胶乳或由KOH保存的NR胶乳,因不含氨而不形成锌络盐,所以很难产生不稳定化,但在高温下连续加热时,在聚合物与浆液、吸附物质与浆液之间会产生反应,结果或者pH值降低,或者出现不稳定化。这也是广义的热稳定性。
另外进行热敏配合的胶乳对温度更加敏感,在低温下较稳定,黏度的经时变化也小,但在某温度以上时会瞬时进行胶凝化。将加入不同温度热水的试管浸渍一定时间(如5~10s)也可以评价测定的最低放凝化温度或者将加入70℃或90℃热水的试管浸渍一定时间时的凝胶附着量或凝胶强度。
5.3.1.12 其他稳定性试验
(1)储存稳定性[13]胶乳在储存中产生物理或化学变化的难度称为胶乳的储存稳定性或存放稳定性。胶乳有时往往储存一年以上,所以储存稳定性是胶乳非常重要的性质。影响胶乳储存稳定性的因素有物理因素和化学因素。物理因素是因粒子与水的密度不同引起的沉降或上浮。而化学因素有脱氯化氢引起的变化或NR中非橡胶成分腐败等。作为观察化学变化的储存稳定性的促进试验,有时进行高温储存试验.
(2)冻结解冻稳定性性[17,18]储存在寒冷地方的胶乳有时往往反复冻结解冻,那时胶乳的分散状态产生变化,解冻后或者黏度上升或者稳定性降低。在极端场合整体凝固而不能恢复原状。对于这种冻结解冻的稳定性称为冻结解冻稳定性或冻结解冻熔解性,更简单地说也称为冻结稳定性,在反复进行冻结温度、时间和冻结解冻循环之后测定黏度或稳定性。对于NR胶乳没有制定标准。但对树脂乳液和合成橡胶胶乳的冻结解冻试验方法作为ISO 1147进行了标准化。
5.3.2 原料胶乳特有的试验方法
NR胶乳除了橡胶烃外还含有其他成分,而且在采集阶段可能有其他成分混入。另外根据储存状态,发现腐败对质量影响很大。因此,对于NR胶乳有许多试验方法。此外作为NR胶乳制品的标准在ISO 2004中规定了氨保存离心浓缩胶乳和膏化浓缩胶乳的标准,以及在ISO 2027中规定了蒸发法浓缩胶乳的标准。
5.1.2.1 橡胶固形物含量物定
刚刚采集的NR胶乳除了橡胶烃外还含有百分之几的非橡胶分。因此胶乳浓缩精制后也含有1%~2%左右的非橡胶分。所以橡胶固形物含量的测定在了解实际的橡胶含量和非橡胶含量方面是重要的测定项目。该橡胶固形物含量测定方法以ISO 126进行了标准化。
橡胶固形物含量的测定由以下方法进行。即,将(10±1)g胶乳(准确至1mg)稀释至总固形物含量为20%,然后在稀释的胶乳中加入酸。
对于用氨作保存剂的胶乳,加入(75±5)cm3的20g/dm3醋酸水溶液进行凝固。将凝固的橡胶在醋酸中溶液中稍微压碎,15min后由蒸汽浴加热30min。在浆液呈白色混浊时加入5cm3乙醇进行凝固。用玻璃棒充分搅拌后,和氨保存胶乳一样将凝固的橡胶在醋酸水溶液中稍微压碎,15min后用蒸汽浴加热30min使其完全凝固。
将加热后橡胶凝固物收集起来洗涤至不显示酸性,然后压碎成2mm以下的薄片。对于氨保存胶乳用水洗涤2h以上,除去水分后在(70±2)℃的恒温箱中进行干燥。最后几小时反复几次充分干燥两面。而后继续干燥,将30min干燥后的质量变成1mg以下的点作为结束。测定结果用干燥后的橡胶质量除以试样质量的百分率表示。
5.3.2.2 非橡胶组分含量测定
非橡胶组分含量可由总固形物含量与干胶含量之差表示。通常离心浓缩胶乳的非橡胶组分含量的值是1.5%~2.0%左右。对于进行两次离心浓缩的胶乳,其非橡胶组分含量值是0.5%左右。该非橡胶组分含量对最终产品的吸水性或硫化速率以及耐老化性均有影响,因此是重要的测定项目。
5.3.2.3 残渣含量测定
残渣含量的测定在ISO 2005中进行了规定。残渣的主要成分是砂、土、树皮和磷酸铵、磷酸镁等沉淀物。测定方法是,由12000m/s2的离心力对胶乳进行离心分离,然后用氨醇混合液(氨水“试剂0.9g/cm3”l0cm3、乙醇“95容量试剂”340cm3、蒸馏水1000cm3使沉淀的残渣再分散,继之反应进行离心分离,将最后残余的沉淀物于(70±2)℃下进行干燥。测定结果由最后残余沉淀物对试验质量的百分率表示。
5.3.2.4 氮含量测定
氮含量与NR中的蛋白质含量有关系,NR固体橡胶和NR胶乳干胶由凯氏法(Kjeldahl's)测定氮含量在ISO 1656中进行了规定。
测定方法是,首先对胶乳进行凝固、干燥,将所得固态橡胶切碎,用硫酸、硫酸钾、催化剂混合物进行分解,使橡胶中的氮化物作为硫酸铵进行固定化,接着在该溶液中加入强碱,蒸馏游离的氨,再由已知当量浓度的酸溶液吸收。然后用碱中和酸溶液,得到所吸收的氨量。最后由该氨量计算橡胶中所含的氮量。
5.3.2.5 铜、锰、铁含量测定
铜、锰、铁对胶乳中的橡胶成分有氧化催化作用。因此,在非橡胶成分含量试验中,对这些金属含量测定是控制橡胶质量的重要的试验项目。特别对于铜、锰,在ISO 2004(离心分离法和膏化法浓缩氨保存胶乳产品标准)和ISO 2027(蒸发法浓缩胶乳产品标准)中规定了铜、锰含量的上限值。这两件标准规定的锰、铜标准值对于胶乳的固形物含量都是8mg/kg。
对于锰含量测定,首先除去影响测定的氯、硅酸盐,然后用过碘化钠氧化除去氯、硅酸盐的橡胶分解物,对锰氧化物的浓度进行比色定量分析。对于铜含量测定,在溶液中使二乙基二硫代氨基甲酸与通过处理除去橡胶燃烧后的残余物或钾、铁的氧化分解橡胶进行反应,然后对其生成物进行比色定量分析。
5.3.2.6 硼酸含量测定
NR的LA(低氨)胶乳是作为二次保存剂添加的,但因硼酸成分能提高后述的氢氧化钾值(KOH No.),因此硼酸含量测定也是修正氢氧化钾值的试验方法。该硼酸含量测定方法在ISO 1802中进行了规定。
测定方法是用300cm3烧杯取试样约10g(准确至0.1g),然后加入非离子表面活性剂5%溶液2cm3和水50cm3,进行充分搅拌。接着一滴一滴地加入2%盐酸,将pH值调整为2.5~5.5,放置15min后再按规定的方法加入标准0.05N的KOH溶掖,充分搅拌后便pH值调整为7.5,加入甘露糖醇4g。由此pH值降低,所以再次加入KOH溶液,读出使pH值保持在7.5上所需KOH溶液的容积(v),再由下式计算硼酸含量。即:
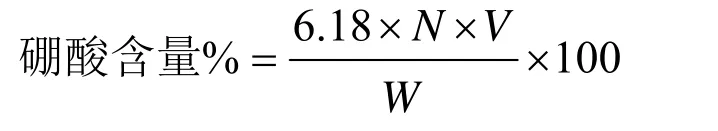
式中N—KOH溶液当量浓度;
W—试样胶乳质量,g。
5.3.2.7 总碱度测定
总碱度测定在ISO 125中做了规定。
测定方法是由200mL蒸馏水稀释称重的胶乳5~10g,由酸中和进行。作为中和使用的酸,规定了0.05mol/dm3的硫酸和0.1mol的盐酸。另外作为中和的终点,可规定为由pH计测定pH值为6.0时,或者以甲基红为指示剂在测定试样变成粉红色时。
测定结果用NR胶乳中的总游离碱量对胶乳的百分率表示。对于氨保存胶乳的系数规定,用硫酸时为3.4,用盐酸时为1.70;对于KOH胶乳的系数规定,用盐酸时为5.61,用硫酸时为11.22。其计算式如下:

式中F—系数;
c—酸浓度,mol/dm3
V—酸添加量,cm3;
m—试样质量,g。
关于氨保存胶乳的测定结果,结果在0.5以上若两次的测定值之差为0.02以上,以及结果在0.5以下若两次的测定值之差为0.0l以上,则再测定—次。另外对于以KOH为保存剂使用的胶乳,两次测定值的差在0.03以上时再测定一次。
5.3.2.8 氧氧化钾值测定
氢氧化钾值(KOH No.)测定是检验NR胶乳保管中其蛋白质腐败程度的一种重要的方法。ISO 127对这种测定方法进行了规定。氢氧化钾值的定义是能中和含100g总固形物NR胶乳生成的铵盐的酸所需当量KOH的克数。因此,测定根据滴定曲线的拐点求出置换酸的铵盐的氨所需KOH量。氨浓度高时拐点难辨认,因此根据碱度和固形物含量加福尔马林溶液,将胶乳中的氨量调整至0.5%后再行测定。此外对于以硼酸为保存剂使用的NR胶乳,预先测定硼酸量修正测定结果。
氢氧化钾值被认为是判断NR胶乳质量的因素,但因为胶乳中所含的酸都能提高氢氧化钾值,所以作为上述二次保存剂加入的硼酸、能提高稳定性的高级脂肪酸和能降低稳定性的挥发性脂肪酸或蛋白质水解产物的氨基酸等也与氢氧化钾有关系。因此,作为稳定性的大致标准,下述挥发性脂肪酸值(VFA No.)被视为重要指标。
5.3.2.9 挥发性脂肪算值
挥发性脂肪酸值(VFA No.)的测定方法在ISO 506中做了规定,可由中和挥发性脂肪酸(VFA)所需KOH量表示。
测定方法如下。即,在称取的胶乳中加入等量的30%硫酸铵,然后加热凝固。由过滤分离凝固物,接着挤出凝固物中所含的浆液,和滤液一起进行测定。在该滤液中加入硫酸进行酸化。此时,KOH因产生沉淀由过滤除掉。用Markham蒸馏装置对酸化的滤液进行蒸馏。用不含二氧化碳的空气对馏出物处理3min,然后以溴百里酚蓝(BTB)或酚酞为示剂由氢氧化钡进行滴淀,算出挥发性脂肪酸量。
5.3.2.10 高速旋转搅拌式机械稳定度试验方法
NR胶乳的机械稳定度试验方法。以ISO 35实行了标准化。该试验方法通常称为MST试验,用具有如图5-3-2所示结构的所谓克拉克逊(Clarkson)式稳定度试验机进行测定。试验是以14000r/min的高速搅拌胶乳,以胶乳中产生凝固物时的时间(s)表示稳定度。
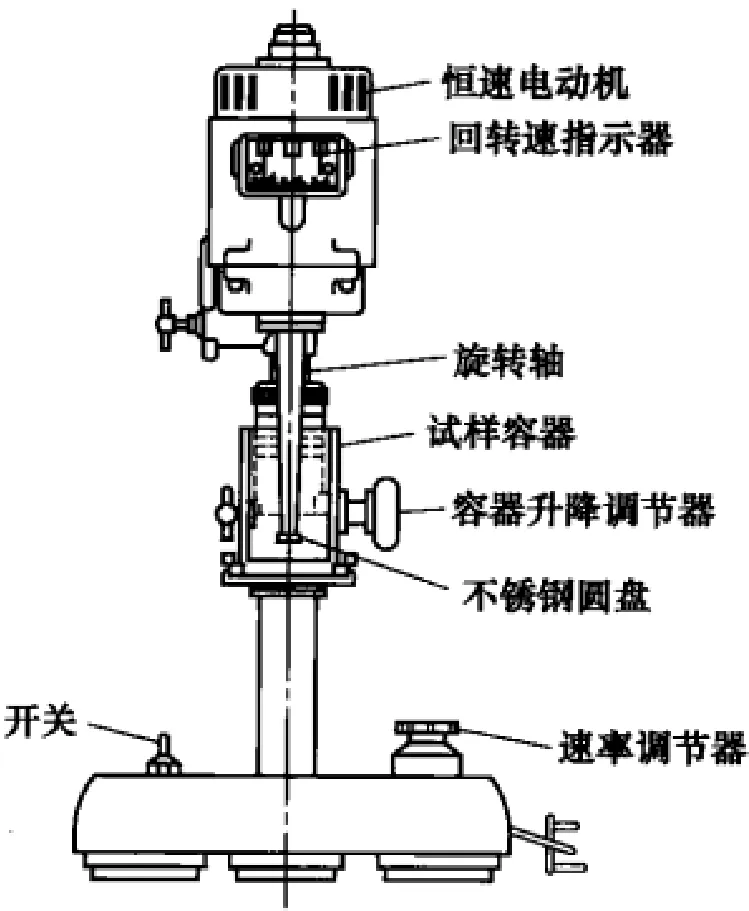
图5-3-2 高速旋转搅拌式(Clarkson式)机械稳定度试验机
试样胶乳在保存中不能冷却。另外试样容器拆封后试验应在24h内进行,在空气中暴露的时间长时机械稳定性可能发生变化。首先对高氨(HA)胶乳用1.6%氨水稀释,对低氨胶乳(LA)用0.6%氨水稀释,总固含量规定为(55.0±0.2)%,边缓慢搅拌边调温至36~37℃。然后立刻用177μm的不锈钢网过滤,在试验容器中称取(80±0.5)g试验胶乳。确定胶乳温度在(35±1)℃,将容器固定在设定的位置上。接着打开电开关,按动秒表,操作速率调节旋钮,尽快调至14000r/min转数。接近终点时试样液面下降,声音变化。为了判断终点,用取样棒取一滴试验中的试样滴在玻璃器皿的水面上,此时若有凝固粒则可分辨滴落和漂浮。最好是能确认在摇动玻璃器皿和吹气振动水面时凝固粒也不散开。在接近终点以15s的间隔进行这种操作,确认凝固粒的产生。稳定度由最初发现凝固物时的秒数表示。试验进行2次。若两次测定结果不超过二次平均值的2.5%,则取其平均值作为该试样的机械稳定度。
5.3.2.11 胶乳的色、味测定
关于NR胶乳的色和味,ISO 2027标准(蒸发浓缩法NR胶乳)没有特别规定试验方法,规定为乳白色,应不显著呈蓝色或灰色,以及由硼酸中和氨后应无腐臭味。但是,关于色和味的定量试验方法没有特别规定。
在以前制定的BSl672(1972)中,关于胶乳气味规定了以下试验方法。即,在125cm3的45℃热水中溶入8g硼酸(总碱度在1.0%以下时为4g),再向其中添加100g胶乳进行充分混合,然后极快移入口径40mm而容积约250cm3的玻璃瓶中,盖上瓶盖放在球磨机台架样的两个辊上,以25~150rpm的速率转动30min。然后取下瓶盖立刻闻味判断胶乳的气味。正常胶乳无腐败臭味,而表现太甜的气味。
5.3.2.12 过滤性测定
NR胶乳凝块含量或淤渣含量尽管是一定的,有时过滤性也显著不同。该过滤性关系到操作性,是NR胶乳的重要性能。布拉斯毛毯过滤性测定方法是将固定有标准毛毯的过滤性测定装置安装在已知质量的吸滤烧瓶上,用油酸钾溶液弄湿,注胶乳进行减压过滤。胶乳滴下的间隔超过5s时规定为终点,然后恢复到常压卸下过滤装置,称烧瓶质量。过滤性由单位面积毛毯过滤的胶乳质量表示。
5.3.2 原料合成胶乳特有的试验方法
在SR胶乳中,由于合成时作为乳化剂多添加表面活性剂,而且残存末反应单体,因此规定了特有的试验方法.
5.3.3.1 皂含量测定
在旧版JISK 6387(SBR胶乳)中规定了可适用于SBR胶乳中脂肪族类和松香酸类单独或者混合物情况的试验方法。该方法使用的表面活性剂种类是已知的,对于使用合成表面活性剂的胶乳多数受不能适用等的限制,缺乏通用性。另外,ISO中也没有规定SR胶乳的皂含量测定方法,因此在1999年重新修订JIS时该试验方法自行废止。
在此只对测定原理简要介绍。首先用盐酸使胶乳呈酸性,使皂含量变成酸的形式。然后用标准苛性钠溶液滴定。按pH值的变化点求出中和值,再按皂当量计算皂含量。
5.3.3.2 残余苯乙烯含量测定
近年随着环保规则的强化,SBR胶乳中所含的残余苯乙烯量也有几个ppm(×10-6)产品。这种极微量的测定用以前的碘滴定法不能解决。
ISO 13741—1,2中规定的测定方法采用了能够分析极微量而且迅速以及精度也高的气相色谱法。柱使用毛细管引入试样,根据直接注入液体法(ISO 13741—1)和液上气体分析法(ISO 13741—2)进行选择。
定量分析法是在试样中添加已知量的内部标准物质,根据测定的色谱内部标准和目标物质的峰面积比求出目标物质的含量。必须预先测定在已知目标物质标准试样中添加了内部标准溶液的色谱,制作校正曲线。
直接液体注入法是将由含内部标准的水进行稀释的试样直接注入气相色谱仪的方法。另外,液上气体分析法是,试样由含内部标准和聚合阻聚剂(2,6-二叔丁基-4-甲基苯酚)的水及亲水性溶液稀释后封入管式瓶中,使管式瓶呈热平衡状态,将液上气体引入气相色谱仪的方法。
5.3.3.3 残余丙烯腈含量测定
在已经废止的JIS K6392(NBR胶乳试验方法)中没有规定这种试验。但是,和残余苯乙烯一样,随着环保规则的加强,残余丙烯腈的削减也成为重要课题。
基于ISO的测定法,和残余苯乙烯含量测定一样,在ISO 13741—1,2中进行了规定,可由相同试验装置、相同试验方法测定。
5.3.3.4 结合苯乙烯含量测定
SBR胶乳的结合苯乙烯含量的测定很早以来就便用折射率法。ISO 3136也以同法规定了结合苯乙烯含量的测定方法。但是,ISO规定的测定方法对羧基改性SBR和VP改性SBR不适宜。
ISO 3136中规定的测定法是,山甲醇/硫酸对胶乳进行凝固,对生成的颗粒进行干燥。然后使聚合物片化,用乙醇—甲苯共沸物抽出,干燥后压制成聚合物片。而后测定该聚合物片的折射率,根据换算表求出结合苯乙烯含量。
结合苯乙烯含量的求取方法除了折射率法以外,还报道过红外线吸收光谱法和热解气相色谱法等各种方法。
5.3.3.5 结合丙烯腈含量测定
NBR胶乳结合丙烯腈含量可根据聚合物中的氮含量求出。
IOS 3900中规定的测定方法是,在70℃下制作放乳薄膜,由水抽出后用凯氏定氮法求出氮含量。该方法也可用于测定羧较基改性NBR胶乳或丙烯腈—异戊二烯胶乳等。
5.3.3.6 氯丁橡胶胶乳残余碱含量测定
氯丁橡胶胶乳在储存中从聚合物中脱离出盐酸与胶乳中的碱进行反应生成盐。因此,游离的残余碱被消耗,结果pH值降低,胶乳不稳定。残余碱含量越少,胶乳越不稳定。
ISO 13773中规定的测定法是由0.1mol/dm3盐酸滴定添加烷基苯酚聚氧乙烯缩合型的10%非离子型表面活性剂进行稳定的胶乳,求出第一和第二拐点,并求出残余碱含量、δ-滴定度和碱度3个值。
5.3.3.7 机械稳定性测定方法(高速法)
该试验方法使用的装置虽然和NR胶乳机械稳定度测定方法相同,但在转动圆盘的尺寸和稳定度表示方法方面不同。该测定方法在ISO 2006中进行了规定。与NR胶乳不同,转动圆盘增大到(36.12±0.03)mm。黏度在200mPa·s(ISO的规定)以上或在350mPa·s(ASTM的规定)以上时,在总固形物不降低10%以上的范围内。用水稀释使该黏度降低,并且将温度调至(25±3)℃,用(180±15)μm的不锈钢网进行过滤,用试验容器称取(50±0.5)g。然后将容器固定在设定的位置上,以(14000±200)r/min搅拌l~30min(ASTM的规定是30min)。此时必须控制胶乳不能达到60℃以上,而且胶乳在容器中的高度不能超过100mm。如果在100mm以上,在容器的上壁部内表面上涂覆膏状有机硅消泡剂,防止增加气泡。然后卸下容器称重。用180μm不锈钢网过滤试样,分离收集凝固物。附着在转动圆盘轴上的凝固物也用5%油酸钾溶液(进行凝固的场合用5%合成阴离于或非离子活性剂溶掖)洗涤加在不锈钢网上。
然后再用5%油酸钾洗涤至没有胶乳,最后用蒸馏水漂洗,于(100±2)℃下进行干燥。机械稳定度用对于胶乳或试验时总固含量的凝固率%表示。在试验结果中,必须记录试验时的总固含量和搅拌时间。
5.3.3.8 低速转动摩擦式机械稳定度试驻方法
该试验方法是使用马朗式(Maron)稳定度试验机的方法。以前,以JIS K6387(SBR胶乳试验方法)和JIS K6329(NBR胶乳试验方法)作为标准发行,但自JIS统合和废除以后,该测定方法没有作为ISO标准或国家标准存在。但是,作为SR胶乳机械稳定度试验方法,考虑与实际情况一致而在JIS废除时作为技术资料保存.
该试验方法使用具有如图5-3-3所示结构的装置,以定负荷将转动圆盘压紧在聚乙烯垫上,以1000r/min转动一定时间进行测定,测定结果用对于试样胶乳固形物含量的凝固率%表示。用180μm的不锈钢网过滤温度调至35℃的胶乳,然后用试样容器称取(50±1)g。将试样容器固定在装置上后放下转动圆盘,对聚乙烯垫表面轻轻挤压,施加规定载荷(5kg、l0kg或15kg)进行固定,然后开始转动。转动一定时间(5min、10min或规定的适宜时间)后停止,测量试样温度,提升转动圆盘,将黏附的试样和试样容器中的试样由蒸馏水通过180μm的不锈钢网进行流洗,然后收集产生的凝固物。此时,干燥生成的胶膜除外。接着分解试样容器,使聚乙烯垫与底盘间存在的胶乳也在不锈钢网上流动,对凝固物进行完全收集。最后充分洗涤至完全是凝固的胶乳,在70~105℃下进行干燥、称量。试验进行3次,试验结果若不离开其平均值20%以上,则用其平均值作为该试样的机械稳定度。在测定结果中,必须记录试验时的负荷、转动时间、上升温度和凝固物形状。
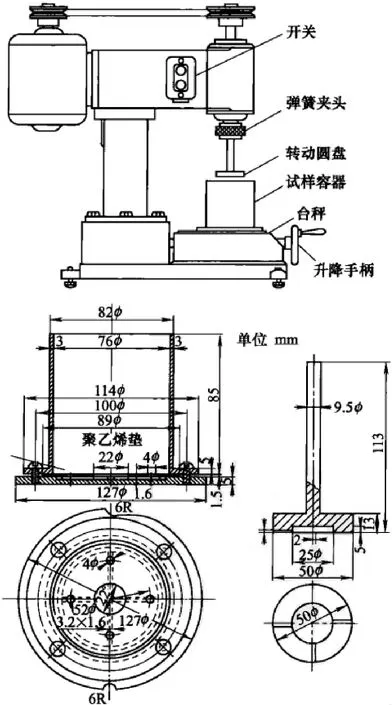
图5-3-3 低速转动摩擦式机械稳定度试验机
5.3.4 配合胶乳试验方法
配合胶乳的试验方法ISO没有作为标准进行特别规定。配合胶乳的试验与原料胶乳相比,因组成不一样其行为也复杂,试验结果评价也多为困难,以及评价方法也依配合胶乳的用途而异,因此作为通用方法可标准化的有限定范围。但是,配合胶乳性能的控制对日常的工艺管理和产品质量管理非常重要。因此,将适当的试验方法标准化是必要的,对此日本橡胶工业技术委员会胶乳分会制定了以下标准。
SRIS 3201 配合胶乳的干胶膜和硫化胶膜的标准制备方法
SRIS 3202 配合胶乳的总固形物含量、pH值、黏度的测定和预硫化判断方法
SRIS 3203 配合胶乳的机械稳定度试验方法
关于配合胶乳的试验方法,下面对预硫化程度和湿凝胶强度进行说明。
5.3.4.1 预硫化程度试验方法
(1)氯仿试验 用不同的烧杯分别取试样胶乳和同容量氯仿,立刻混合后山玻璃棒搅拌至完全凝固。一旦凝固就原封不动放置30~60s,然后用双手拿着橡胶块拉伸,根据其伸长情况、断裂方向、切口的黏合性和形成一体的程度等将预硫化进行程度分为4个阶段。
第一阶段 是几乎没有进行预硫化的阶段,黏性大,整体呈年糕状凝固块,拉伸时不费很大力就能拉得相当长。
第二阶段 拉伸长好像感觉有相当的阻力,但还有伸长或黏性,当挤压切口时黏合成一块。湿凝胶强度处于更高状态,是良好的成熟期。
第三阶段 拉伸时伸长不太大即断裂,变得几乎无黏性,而且即使对切口挤压也不黏合。预硫化虽然增进相当大,但对加工还没有造成显著障碍。但是由于湿凝胶强度降低,必须注意加工方法。
第四阶段 进行凝固但因黏性不足也设有变成一个统一的小块。预硫化进一步加强而变成细粉末状。
该试验方法虽然不是定量分析,但操作简单可迅速进行。因此熟练操作时可正确地判断预硫化程度。特别对于生产时预硫化状态的确定,由于试验中不使用费时间的试验方法,所以这是一种有效的方法。
(2)溶胀直径试验 测定由溶剂引起的平衡溶胀率,是判断预硫化程度的定量试验方法。试验精度比氯仿试验还优异,但试验时间稍长一些。用试样胶乳通过淹延或浸渍,制作0.1~0.2mm厚度的干胶膜。此时立即吹风,于室温下(35℃以下)进行干燥。为防止黏合用滑石粉进行无残余变形剥离,用带红色标记油墨的刀具冲切成直径15mm的圆形试样。然后将装满二甲苯的陪替氏皿放在方格纸上,用镊子将冲切的试样沉入二甲苯中。溶胀率按达到平衡溶胀时进行正交的两直径尺寸和冲切直径由下式计算:
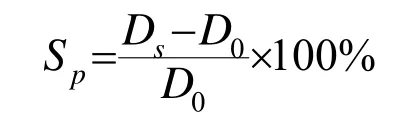
式中Sp—溶胀串,%;
Ds—平衡溶胀时的试样直径,mm;
D0—冲切时的试样直径,mm。
按配合后未硫化胶乳和对完全硫化试样的测定值由下式计算预硫化程度:
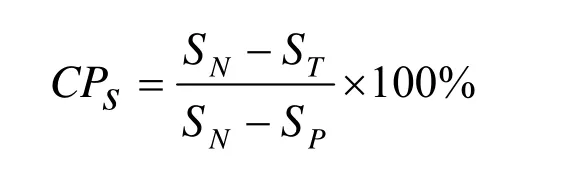
式中CPs—溶胀法的预硫化程度,%;
SN—来硫化试样溶胀率的平均值,%;
SP—完全硫化试样溶胀率的平均值,%;
ST—预硫化试样溶胀率的平均值,%。
(3)永久变形试验 根据永久变形也可以判断预硫化程度。但是,为了均匀伸长需要比上述溶胀直径试验还厚的胶膜,对于测定需要稍长时间。试验方法按JIS K6301的测定方法求出永久变形,预硫化程度的计算按上述方法进行。用于试验的试样尺寸为宽6mm、长112mm(平行部分为100mm)。
5.3.4.2 湿凝胶强度试验
湿凝胶强度对于加工性是重要的因素,虽然进行过多种试验,但没有标准化方法。湿凝胶试样的制备方法有以下几种:①对浸渍凝固剂的薄页纸进行浸渍制作湿凝胶的方法;②使配有铵盐的胶乳在环形模具中进行胶凝化的方法;③使硅氟化钠配合胶乳在哑铃状模具中进行胶凝化的方法;④用组合模具浇注圆柱形式样的方法等。但由于湿凝胶的结构从形成起,时刻在变化,强度随着时间进行变化,所以在考虑数据波动时最好是在脱水收缩处于平衡状态的时候进行。
作为测定方法,在此记录以SRIS草案研究的一例。首先用50%硝酸钙(四水合盐)甲醇溶液由凝固浸渍法制作厚度约1mm的湿凝胶,然后在50%醋酸水溶液中浸渍3~5min,水洗后夹在玻璃纸中由JIS 2号刀具冲切试样,储存在一定温度水中,取出后立即进行拉伸试验。
若能追踪脱水收缩达到平衡状态以前的变化过程,效果可能会更好。为了测定胶丝制造过程的凝胶强度,也在研究使用由喷嘴挤出到凝固浴中的线状温凝胶,用经时进行计测的方法或用扭力试验仪对胶乳的凝胶化过程进行追踪的方法。
1)ISO/TC 45国内審議委員会:ISO/TC45国内審議委員会新JIS資料,日本ゴム工業会(1999)p.1.
2) Philpott. M.W. Sekar, K C ∶Journal of the Rubber Research Institute of Malaya, 14, 93 (1953).
3) Maron, S.H., Ulevltch, I.N., Elder. M.E.∶ Analytical Chemistry 24, 1068 (1952
4) Lucas, F.F.∶ Industrial and Enginnering Chemistry, 30, 146 (I938).
5) Schoon. Th. G F, Phoa. K L∶ Journal of Collids Science, 10. 26 (1955).
6) Maron, S. H. Eider. M, E, Ulevltch, 1. N∶ Jotcrnal of Collids Solace, 9. 89 (1954).
7) Kubitschek, H.E∶ Research, 13. 128 (1960).
8) Matthews, B.A, Rhodes. C.T∶ Journal of Colloid and Intcrface Science, 28. 71 (1968).
9) Blackey. D.C., Ong. E K∶ Journal of the Institution of the Rubber Industry, 4(1), 17 (1970),
10) Mutphy, E. A.∶ Proceedings of tke Rubber Technology Conference, London (1938)p. 151,
11) Doeson, H.G.∶ Rubber World, 135, 239 (I956).
12) Newnham, J. L. M. Simcox, D. J.∶ Proceedings of the Rubber Technology Conference, Washington (1959) p 323.
13) 室井宗一:高分子ラテツクスの化学,高分子刊行会(1970)p.176
14) 室井宗一:高分子ラテツクスの化学,高分子刊行会(1970)p.219
15) Blackley, D. C∶ Polymer Latices Vol. 1 (2 nd ed.). Chapman & Hall London (1997) p. 187,
16) ISO/TC 45国内審議委員会:ISO/TC45国内審議委員会新JIS資料,日本ゴム工業会(1999)p.23.
17) Maron, S H, Ulevitch. I. N. Analytical Chemistry, 25. 1087 (1953).
18) ゴム技術調查研究報吿書,日本ゴム協会(1970)p.59.
(未完待续)
10.3969/j.cn.12-1350(tq).2014.05.006
猜你喜欢
辽宁化工(2024年6期)2024-07-11 19:02:07
四川蚕业(2022年2期)2022-11-19 02:10:14
玩具世界(2022年1期)2022-06-05 07:42:30
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
中国民航大学学报(2021年2期)2021-06-24 01:08:16
橡胶工业(2018年11期)2018-07-23 07:34:10
石油沥青(2018年3期)2018-07-14 02:19:18
国际木业(2016年8期)2017-01-15 13:55:09
橡塑资源利用(2015年1期)2015-12-24 06:29:17
橡胶工业(2015年4期)2015-07-29 09:17:08
