
松香基季铵盐为模板剂有序超微孔二氧化硅的合成
2015-12-15 07:18陈尚钘赵振东王宗德范国荣
无机化学学报 2015年4期
王 鹏 陈尚钘*, 黄 敏 赵振东 王宗德 范国荣
(1江西农业大学林学院,南昌330045)
(2中国林业科学研究院林产化学工业研究所,南京210042)
松香基季铵盐为模板剂有序超微孔二氧化硅的合成
王 鹏1陈尚钘*,1黄 敏1赵振东2王宗德1范国荣1
(1江西农业大学林学院,南昌330045)
(2中国林业科学研究院林产化学工业研究所,南京210042)
以松香基季铵盐(脱氢枞基三甲基溴化铵,标记为DTAB)为模板剂、正硅酸乙酯为硅源、氨水为碱性介质成功合成出具有纳米片状形貌的六方有序超微孔二氧化硅材料。采用X射线衍射、N2吸附-脱附、透射电镜、扫描电镜等手段对样品进行表征,结果表明,体系中模板剂添加量、硅源添加量、碱性介质添加量、晶化温度、搅拌时间对前驱体的有序度有着较大的影响。当物质的量之比为nSiO2∶nDTAB∶nNH3·H2O∶nH2O=1.0∶0.1∶11.3∶924.0,晶化温度为373 K,搅拌时间为24 h,所得样品有序度最高。经煅烧后样品具有较大的比表面积(1 024 m2·g-1)和孔容(0.56 cm3·g-1),以及狭窄的孔径分布(集中于1.80 nm)。
松香基季铵盐;有序超微孔;二氧化硅
由于在化学化工、生物医药、光电器件、能源环境等领域的广阔前景[1-4],有序介孔材料一经问世便备受关注[5-6]。广大科研人员通过“软模板”法合成出多系列不同结构的有序介孔材料,例如:SBA[7-9], HMS[10],MSU[11]和FDU[12-13]等。这些材料突破了微孔材料1.2 nm的束缚,将有序多孔材料孔径可达到的范围扩展至2.7~12 nm。人们不断尝试扩大有序介孔材料的孔径以拓展其应用领域[14-15],却忽略了其孔径的缩小,即有序超微孔材料的合成。事实上,作为孔径介于微孔(Dp<1.2 nm)与介孔(Dp>2.7 nm)之间的一类特殊多孔材料,有序超微孔材料有着重要的用途。例如在重油加工、新药中间体合成、特殊化学品生产中,传统微孔分子筛孔径较小,大分子化合物难以进入其孔道,而有序介孔分子筛的孔径则过大,对催化产物难以实现择形功能,有序超微孔材料则成为解决上述问题的理想材料[16-17]。此外,在特定尺寸化合物分离以及难溶药物增溶与缓释等领域,有序超微孔材料也具有其它多孔材料无法比拟的优势[18]。
有序超微孔材料的合成方法与有序介孔材料类似,主要利用表面活性剂的自组装体系为模板,即“软模板”法来合成[3-4]。但有序超微孔材料处于狭窄而特殊的孔径区间,对表面活性剂的要求更为苛刻,由于有序介孔材料(M41S)的孔径大小与所用模板剂的疏水链长短有关,人们开始尝试采用短链表面活性剂合成有序超微孔材料,但短链表面活性剂在水中形成胶束能力较弱,难以与无机物种自组装形成有序体系,如果以单一传统短链表面活性剂为模板剂,所得材料往往有序度较差或为无序蠕虫状结构[19-21]。为了解决这个问题,人们合成了系列具有特殊结构的两亲模板剂(包括端位具羟基的Bola型表面活性剂[16,22],双短链Gemini型表面活性剂[23-24],离子液体[25-26],半氟化表面活性剂[27]等),并应用于有序超微孔材料的合成。研究表明,模板剂的分子结构对所合成有序超微孔材料的结构性能起着决定性的作用。因此,探索和开发具有新结构的模板剂已经成为有序超微孔材料开拓创新的必经之路。
在本研究中,我们以松香基季铵盐为模板剂合成有序超微孔材料,与传统直链基表面活性剂不同,松香基表面活性剂具有独特的三环菲刚性骨架结构,其较强的憎水性增强了其自组装能力,而其较小的分子半径则有利于超微孔材料的形成。本研究重点考察了物质的量之比、晶化温度及搅拌时间对材料结构的影响。
1 实验部分
1.1 药品与仪器
正硅酸乙酯(分析纯):中国医药集团上海化学试剂公司;氨水(分析纯):南京化学试剂有限公司;脱氢枞胺(98%):杭州万景新材料有限公司;甲酸(分析纯):西陇化工股份有限公司;甲醛(分析纯):西陇化工股份有限公司;溴甲烷(化学纯):常州化工研究所有限公司;X射线粉末衍射仪(XRD):Bucker D8 Advanced;全自动物理吸附分析仪:Micromeritics ASAP2020;透射电镜(TEM):JEOL JEM-2100,扫描电镜(SEM):Hitachi S-3400
1.2 实验过程
1.2.1 模板剂的合成
以脱氢枞胺为原料,根据文献[28]以酸醛法合成N,N-二甲基脱氢枞胺,将所得N,N-二甲基脱氢枞胺溶解于丙酮中,按N,N-二甲基脱氢枞胺与溴甲烷的物质的量之比为1∶2的比例,快速加入溴甲烷,密封反应器搅拌过夜。以乙醇-乙酸乙酯重结晶所得产物3次,得模板剂脱氢枞基三甲基溴化铵(标记为DTAB,图1)。
1.2.2 正硅酸乙酯为硅源合成有序超微孔二氧化硅
将碱性介质氨水加入到去离子水中,再加入模板剂,搅拌至溶液澄清,向溶液中缓慢滴加正硅酸乙酯(TEOS),继续搅拌,将所得悬浊液移入内有聚四氟乙烯内衬的不锈钢水热反应釜中,放在鼓风干燥箱中,于一定温度下静置晶化。经特定时间晶化后,取出产物并经抽滤、洗涤、干燥得有序超微孔二氧化硅前驱体粉末。以无水乙醇为溶剂,经索氏提取法去除模板剂,将所得材料放入马弗炉中,以1 K·min-1的升温速率升至823 K煅烧4 h,得到有序超微孔二氧化硅分子筛。
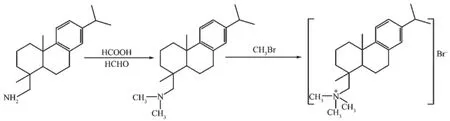
图1 脱氢枞基三甲基溴化铵的合成路线Fig.1 Synthesis route of dehydroabietyltrimethyl ammonium bromide
2 结果与讨论
2.1 DTAB添加量对材料结构的影响
由图2可见,当DTAB添加量较低时,样品仅在2θ=3.01°(d100=2.93 nm)出现1个肩峰(图2a),说明样品并没有形成规整的孔道,可见反应体系中含量较低的DTAB并不足以与硅酸盐物种协同组装形成有序结构。随着DTAB添加量的增加,样品的XRD图出现明显的衍射峰(图2b),说明DTAB添加量的增加有利于样品有序度的提高。当DTAB添加量上升到n SiO2∶nDTAB=1.0∶0.1时,样品在2θ=2.94°、5.11°和5.85°(d100=3.00 nm,d110=1.73 nm,d200=1.51 nm)分别出现3个衍射峰(图2c),这3个峰可归属为二维六方相(P6mm)的100、110和200晶面衍射峰,说明样品已经形成二维六方有序结构。样品主峰的晶面间距为3.00 nm,其值远小于传统有序介孔材料MCM-41主峰的晶面间距,说明所得样品是一种具有较小孔径的六方有序二氧化硅材料。随着DTAB添加量的继续增加,样品的小角衍射峰开始变宽,且峰强度有所下降,说明过高的DTAB添加量会破坏样品孔结构的有序度,这可能是因为当模板剂浓度过高时,模板剂胶束的密度过大,体系中没有足够的空间让硅酸盐物种与模板剂胶束充分接触,从而难以形成规整的孔道。
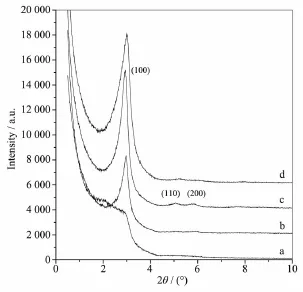
图2 不同DTAB添加量合成二氧化硅前驱体的小角X射线衍射图Fig.2 Small-angle XRD patterns of as-synthesized silicas synthesized with different DTAB dosages
2.2 TEOS添加量对材料结构的影响
由图3可见,当硅源TEOS添加量较小时,样品仅在2θ=2.98°(d100=2.96 nm)和2θ=5.84°(d200=1.51 nm)出现二维六方相(P6mm)100和200晶面的衍射峰(图3a),表明样品已经出现六方有序结构,但有序程度较差。随着硅源添加量的上升,样品开始在2θ= 5.14°(d110=1.72 nm)出现六方相110晶面的的衍射峰(图3b),说明样品有序程度有所提高。当硅源在反应体系中物质的量之比上升到nSiO2∶nDTAB=1.0∶0.1时,样品在100、110和200晶面的衍射峰峰形最尖锐、峰强度最强(图3c),表明此时样品的有序程度最高。当硅源添加量过高时,样品仅在2θ=2.80°(d100=3.15 nm)出现1个肩峰,表明样品已经失去六方有序结构。上述现象表明,适当的硅源添加量有利于样品有序结构的形成,添加量过高或过低均会破坏产物的有序结构。硅源添加量的高低决定着水解后所形成的硅酸盐物种浓度的大小,当硅酸盐物种浓度较小时,其在模板剂上缩聚速率较慢,难以形成良好的孔壁,当硅酸盐物种浓度较大时,缩聚速率过快,造成有序结构的破坏。
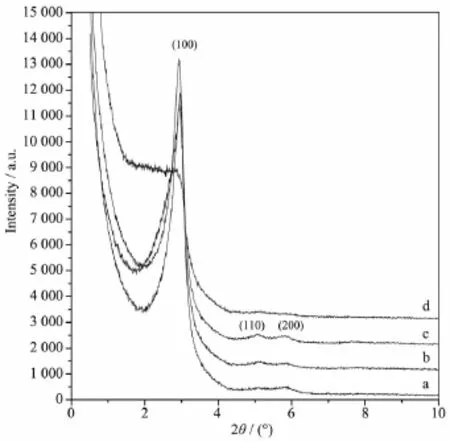
图3 不同TEOS添加量合成二氧化硅前驱体的小角X射线衍射图Fig.3 Small-angle XRD patterns of as-synthesized silicas synthesized with different TEOS dosages
2.3 NH3·H2O添加量对材料结构的影响
由图4可见,当NH3·H2O添加量较小时,样品仅在2θ=2.96°(d100=2.98 nm)出现1个衍射峰(图4a),表明样品并没有形成有序的六方孔道。随着NH3· H2O添加量的增加,体系pH值逐渐上升,样品开始在2θ=2.96°、5.10°和5.79°(d100=2.98 nm、d110=1.73 nm、d200=1.53 nm)分别出现六方相的100、110和200晶面的衍射峰(图4b),说明样品已经形成六方有序结构。当pH值上升至约为10时,样品的3个衍射峰峰形最尖锐,峰强度最强(图4c),说明此时样品的有序度最高。当体系pH值继续上升时,样品的有序度开始下降(图4d),并最终只出现1个衍射峰(图4e)。调控NH3·H2O的添加量实际上就是调控反应体系的pH值,pH值的高低直接决定有机硅源的水解速率,进而影响硅酸盐物种的缩聚速率。较低pH值时,有机硅源水解速率较慢且难以充分水解,体系中硅酸盐物种不能有效发生缩聚形成孔壁,造成产物结构较差,且产率也较低。过高的pH值会使得有机硅源水解过快,硅酸盐物种的缩聚也过快,产物难以形成优良的孔结构。
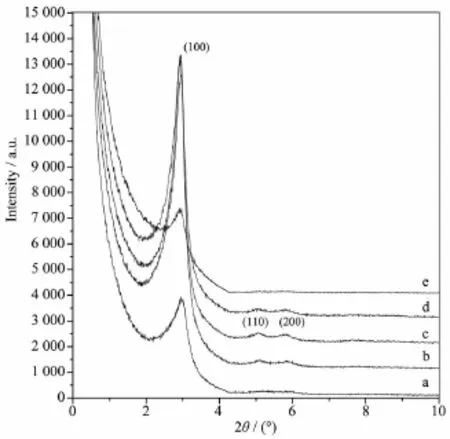
图4 不同NH3·H2O添加量合成二氧化硅前驱体的小角X射线衍射图Fig.4 Small-angle XRD patterns of as-synthesized silicas synthesized with different NH3·H2O dosages
2.4 不同晶化温度对材料结构的影响由图5可见,当晶化温度为303 K时,样品在2θ=3.34°(d100=2.64 nm)出现1个100晶面的衍射峰(图5a),说明样品并没有形成有序结构。当晶化温度升高至343 K时,样品在2θ=3.04°(d100=2.90 nm)出现1个尖锐的衍射峰(图5b),表明随着晶化温度的升高,样品孔道有序度有所提高,当晶化温度升高至363~383 K时,样品出现六方相的100、110和200特征衍射峰(图5c、5d和5e),说明样品已经形成六方有序孔道。衍射峰的尖锐度与峰强度随着晶化温度的升高呈现先上升后下降的趋势。当晶化温度升至393 K时,样品仅在2θ=2.83°(d100=3.12 nm)出现1个衍射峰(图5f),可见更高的晶化温度会造成样品孔道有序度的下降。当晶化温度达到413 K时,样品在2θ=2.04°(d001=4.33 nm)和2θ=3.76°(d002=2.35 nm)出现层状相的001与002晶面衍射峰(图5g),表明样品已经从六方结构开始向层状结构转变。随着晶化温度的升高,样品经过了由无序结构变为六方有序结构,再到无序结构,并最终转化为层状结构的历程。这主要是因为较低的温度不利于硅酸盐物种的晶化,使得样品难以形成有序的硅孔壁,但过高的晶化温度会造成有序体系的扭曲、变形,甚至改变体系的聚集状态,最终形成层状结构。陈晓银等[29]在以十六烷基三甲基溴化铵为模板剂合成MCM-41时发现,晶化温度超过423 K时,样品会由六方结构初步转变为层状结构,而以DTAB为模板剂合成材料过程中,在413 K时,样品已经开始转变为层状结构,说明DTAB胶束的聚集状态对温度更敏感。
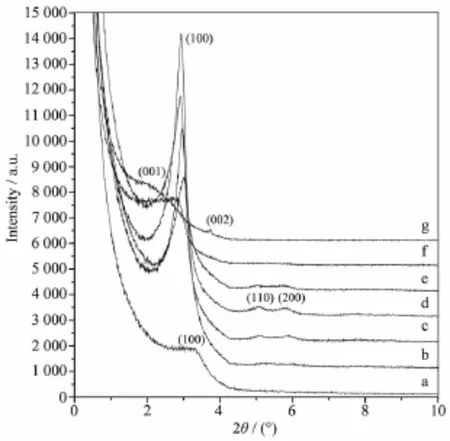
图5 不同晶化温度下二氧化硅前驱体的小角X射线衍射图Fig.5 Small-angle XRD patterns of as-synthesized silica synthesized at different crystallization temperatures
2.5 不同搅拌时间对材料结构的影响
由图6可见,当搅拌时间为0.5 h时,样品仅在2θ=2.96°(d100=2.98 nm)出现1个衍射峰(图6a),说明搅拌时间过短不利于形成高度有序的孔结构。当搅拌时间延长至4~24 h时,样品均出现六方相的100、110和200晶面衍射峰(图6b、6c和6d),其中搅拌时长为24 h时,所得样品的衍射峰峰形最尖锐,峰强度最强,说明样品有序度最高。当搅拌时间超过24 h时,样品在110和200晶面的衍射峰逐渐消失,说明样品有序度开始下降。研究表明,在以十六烷基三甲基溴化铵为模板剂合成MCM-41[5-6]时,搅拌时间只需0.5 h,样品便会形成高度有序的六方介孔结构。这是因为以阳离子表面活性剂合成材料时,表面活性剂与硅酸盐物种以S+I-路线结合并组装成孔壁,两者通过较强静电作用结合,形成孔壁速度较快。本研究虽然是以阳离子表面活性剂为模板剂,应该遵循S+I-路线,但所用脱氢枞基三甲基溴化铵与十六烷基三甲基溴化铵在结构上有着较大的不同,其特有的三环菲骨架疏水基是一种空间位阻较大的基团,该基团的存在会在一定程度上阻碍硅酸盐物种与模板剂胶束的结合,形成孔壁的速率较慢,因此需要较长的搅拌时间才能获得高度有序的孔结构。

图6 不同搅拌时间合成二氧化硅前驱体的小角X射线衍射图Fig.6 Small-angle XRD patterns of as-synthesized silicas synthesized with different stirring times
2.6 煅烧对材料结构的影响
由图7可见,经过823 K煅烧的样品在2θ= 3.06°、5.29°和6.08°(d100=2.88 nm,d110=1.67 nm,d200= 1.45 nm)分别出现六方相的100、110和200晶面衍射峰(图7b),说明样品在823 K煅烧后仍能保持高度有序的六方结构。由于模板剂的清除,煅烧后产物的衍射峰峰形更加尖锐,峰强度也有所增加。相对于未煅烧的前驱体,煅烧后样品的小角X射线衍射峰均向高角度方向有所偏移,这是高温煅烧造成样品晶胞收缩所致。
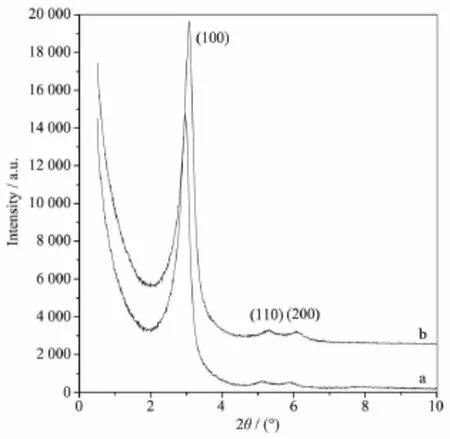
图7 煅烧前后二氧化硅的小角X射线衍射图Fig.7 Small-angle XRD patterns of as-synthesized and calcined silicas
2.7 材料的N2吸附-脱附表征
图8为经煅烧后二氧化硅的N2吸附-脱附曲线,该曲线处于Ⅰ型和Ⅳ型吸附曲线的过渡态[30],说明样品孔径介于微孔与介孔之间,曲线在分压为0.1以下便形成毛细凝聚,说明样品有着较小的孔径。图9为经煅烧后二氧化硅的孔径分布曲线,由于样品孔径处在微介孔材料分界点上,常规HK、BJH模型并不能完整描述所得材料的孔径分布情况,故选用DFT模型进行孔径分布计算。由图9可见,材料有着狭窄的孔径分布,孔径集中分布在1.80 nm处,说明所得样品为有序超微孔材料。样品的BET比表面积为1 024 m2·g-1,孔容为0.56 cm3·g-1。根据样品100晶面间距(d=2.88 nm),通过公式a0=2d/计算样品六方晶胞参数为3.33 nm,减去孔径(1.80 nm)得孔壁厚度为1.53 nm。
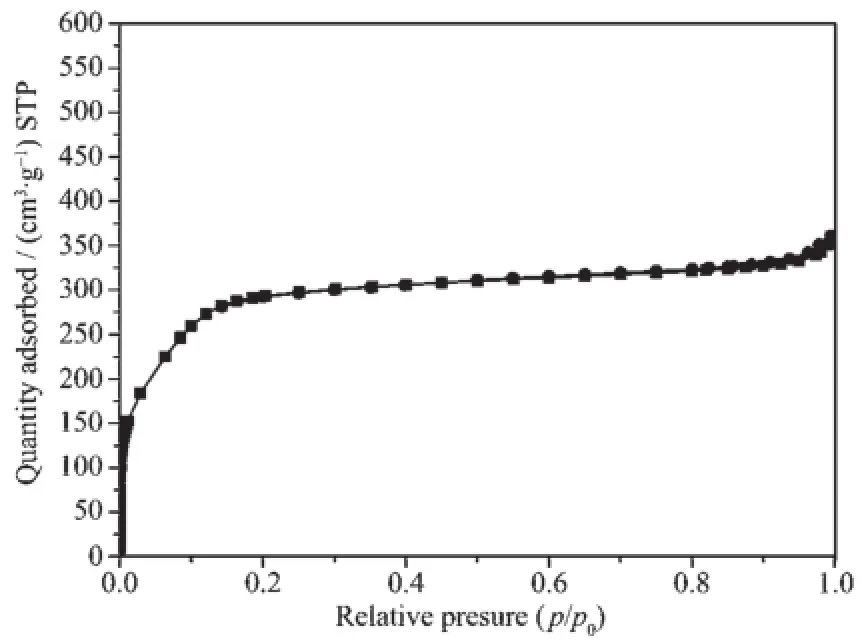
图8 煅烧后二氧化硅的N2吸附-脱附曲线Fig.8 N2adsorption-desorption isotherm of calcined silica
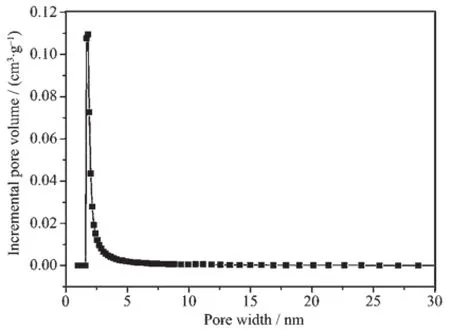
图9 煅烧后二氧化硅的DFT孔径分布曲线Fig.9 DFT pore size distribution of calcined silica
2.8 材料的TEM表征
由图10a可见,在垂直于孔道的方向(001)上,样品显示出长程有序的六方有序超微孔孔道(白色为孔道,黑色为孔壁)。由图10b可见,样品在平行孔道方向(110)上显示出规整有序的超微孔孔道结构。煅烧后样品依然保持着高度有序的六方相结构(图10c和10d),孔径大小大约为1.8 nm左右,孔壁厚度约为1.5 nm,所得结果与N2吸附-脱附表征结果一致。
2.9 材料的SEM表征
为了精确地观测样品的形貌,对煅烧后的有序超微孔二氧化硅进行了扫描电镜表征,由图11可见,样品为纳米片状结构,然而纳米片本身并没有规整的形状,且尺寸也不均一。

图10 煅烧前后二氧化硅的TEM照片Fig.10 TEM images of as-synthesized and calcined silicas

图11 煅烧后二氧化硅的SEM照片Fig.11 SEM image of calcined silica
3 结论
采用松香基季铵盐(脱氢枞基三甲基溴化铵)为模板剂引导合成出有序超微孔二氧化硅材料。体系中模板剂添加量、硅源添加量、碱性介质氨水添加量、晶化温度、搅拌时间对前驱体的有序度起着决定性的作用,当物质的量之比为nSiO2∶nDTAB∶nNH3·H2O∶nH2O=1.0∶0.1∶11.3∶924.0,晶化温度为373 K,搅拌时间为24 h,所得样品有序度最高。经煅烧后,所得样品为六方有序超微孔结构,呈纳米片状形貌,并具有较大的比表面积(1 024 m2·g-1)和孔容(0.56 cm3·g-1),以及狭窄的孔径分布(集中于1.80 nm)。所合成的材料在择形催化、分离以及难溶药物增溶与缓释等领域具有广阔的应用前景。
[1]Corma A.Chem.Rev.,1997,97(6):2373-2420
[2]Charnay C,Bégu S,Tourné-Péteilh C,et al.Eur.J.Pharm. Biopharm.,2004,57(3):533-540
[3]Fan J,Yu C,Gao F,et al.Angew.Chem.,2003,115(27): 3254-3258
[4]Crossland E J,Noel N,Sivaram V,et al.Nature,2013,495 (7440):215-219
[5]Kresge C,Leonowicz M,Roth W,et al.Nature,1992,359 (6397):710-712
[6]Beck J,Vartuli J,Roth W,et al.J.Am.Chem.Soc.,1992, 114(27):10834-10843.
[7]Huo Q,Margolese D I,Stucky G D.Chem.Mater.,1996,8 (5):1147-1160
[8]Yu C,Tian B,Fan J,et al.J.Am.Chem.Soc.,2002,124 (17):4556-4557
[9]Zhao D,Feng J,Huo Q,et al.Science,1998,279(5350):548-552
[10]Tanev P T,Pinnavaia T J.Science,1995,267(5199):865-867
[11]Bagshaw S A,Prouzet E,Pinnavaia T J.Science,1995,269 (5228):1242-1244
[12]Yu C,Yu Y,Zhao D.Chem.Commun.,2000(7):575-576
[13]Liu X,Tian B,Yu C,et al.Angew.Chem.Int.Ed.,2002,41 (20):3876-3878
[14]Fan J,Yu C,Wang L,et al.J.Am.Chem.Soc.,2001,123 (48):12113-12114
[15]Zhou G,Chen Y,Yang J,et al.J.Mater.Chem.,2007,17 (27):2839-2844
[16]Bagshaw S,Hayman A.Adv.Mater.,2001,13(12/13):1011-1013
[17]Davis M E.Nature,2002,417(6891):813-821
[18]Zhao S X,Lu G Q M,Hu X.Chem.Commun.,1999(15): 1391-1392
[19]Serrano D P,Aguado J,Garagorri E.Chem.Commun.,2000 (20):2041-2042
[20]Kruk M,Jaroniec M,Sayari A.J.Phys.Chem.B,1997,101 (4):583-589
[21]Beck J,Vartuli J,Kennedy G,et al.Chem.Mater.,1994,6 (10):1816-1821
[22]Bagshaw S A,Hayman A R.Microporous Mesoporous Mater., 2001,44:81-88
[23]Ryoo R,Park I S,Jun S,et al.J.Am.Chem.Soc.,2001,123 (8):1650-1657
[24]Wang R,Han S,Hou W,et al.J.Phys.Chem.C,2007,111 (29):10955-10958
[25]Zhou Y,Antonietti M.Adv.Mater.,2003,15(17):1452-1455
[26]Zhou Y,Antonietti M.Chem.Mater.,2004,16(3):544-550
[27]Di Y,Meng X,Wang L,et al.Langmuir,2006,22(7):3068-3072
[28]ZENG Tao(曾韬),PENG Shu-Jing(彭淑静).J.Nanjing For.Univ.:Nat.Sci.Ed.(南京林业大学学报:自然科学版), 1996(01):26-28
[29]CHEN Xiao-Yin(陈晓银),DING Guo-Zhong(丁国忠),CHEN Hai-Ying(陈海鹰),et al.Chem.J.Chinese Universities(高等学校化学学报),1997(02):186-189
[30]Brunauer S,Deming L S,Deming W E,et al.J.Am.Chem. Soc.,1940,62(7):1723-1732
Synthesis of Ordered Superm icroporous Silica Using Rosin-Based Quaternary Ammonium Salt
WANG Peng1CHEN Shang-Xing*,1ZHAO Zhen-Dong2WANG Zong-De1FAN Guo-Rong1
(1College of Forestry,Jiangxi Agricultural University,Nanchang 330045,China)
(2Institute of Chemical Industry of Forestry Products,CAF,Nanjing 210042,China)
Ordered hexagonal supermicroporous silica with nanosheet morphology was successfully synthesized using rosin-based quaternary ammonium salt(dehydroabietyltrimethyl ammonium bromine,short for DTAB)as template agent,tetraethyl orthosilicate as silicate source,and ammonia as alkaline medium.XRD,N2adsorptiondesorption,TEM,and SEM were used to characterize the samples.The results indicated that the dosage amount of template agent,silicate source,alkaline medium,crystallization temperature and stirring time had great effects on the regularity of the pore structure.When the mole ratio of nSiO2∶nDTAB∶nNH3·H2O∶nH2Owas 1.0∶0.1∶11.3∶924.0; crystallization temperature was 373 K;stirring time was 24 h,the sample got the hightest regularity.After the calcination,the synthesized material possessed large surface area(1 024 m2·g-1),high pore volume(0.56 cm3·g-1) and narrow pore size distribution(centered at about 1.80 nm).
rosin-based quaternary ammonium salt;supermicroporous;silica
O613.72
A
1001-4861(2015)04-0767-07
10.11862/CJIC.2015.105
2014-11-05。收修改稿日期:2015-01-12。
教育部博士学科点专项科研基金课题(No.20133603120002)资助项目。*
猜你喜欢
保定学院学报(2022年6期)2022-12-01
火炸药学报(2022年5期)2022-11-04
辽宁化工(2022年8期)2022-08-27
建材发展导向(2021年13期)2021-07-28
陶瓷学报(2020年5期)2020-11-09
硅酸盐通报(2020年1期)2020-02-25
陶瓷学报(2019年6期)2019-10-27
天津医科大学学报(2019年3期)2019-08-13
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
