
灰树花子实体转录组测序和分析
2015-11-19 05:50:18袁卫东宋吉玲王伟科
复旦学报(自然科学版) 2015年5期
袁卫东,陆 娜,陈 青,宋吉玲,王伟科
(1.杭州市农业科学研究院,杭州 310024;2.浙江省农业技术推广中心,杭州 310020)
灰树花(Grifola frondosa,maitake)是一种食、药兼用的珍稀蕈菌[1],野生灰树花常生长于栗树周边,俗称“栗蘑”.子实体香气宜人,口感嫩脆鲜美;同时药用价值极高,主要用于消化道、乳腺及前列腺等癌症的治疗,此外也用于血管硬化、血压增高及心脏等疾病的治疗,因此具有良好的开发前景[2].
选育出优质、高产、抗逆性强的灰树花品种,提高其经济效益,一直是灰树花遗传育种工作研究的重点[3].转录组学研究作为一种快速、高通量、全面解读食用真菌的全新技术手段,已经被越来越广泛地应用于食用菌的遗传育种工作中[4].食用菌转录组的研究,可从整体水平上发掘食用菌药用成分生物合成途径中的关键基因,确定有效药用活性成分的合成途径及其调控机制,为食用菌功能基因的挖掘、品种鉴定、资源保护和种质繁育提供新的思路和方法[5].
我们通过对灰树花子实体转录组的测序研究,筛选出灰树花子实体成熟期特异表达基因,并对这些表达的基因进行生物信息学分析,旨在发现灰树花子实体生长过程中的相关基因,为通过基因工程培育优质、高产的灰树花新品种提供理论基础.
1 材料与方法
1.1 试材
供试菌株为灰树花小黑汀,引自山东泰安.菌丝长满菌包后移入出菇大棚培养7~10d,待菌丝扭结形成原基后25d左右形成成熟的灰树花子实体,收集成熟的子实体样品.
1.2 方法
1.2.1 灰树花子实体总RNA 提取及测序
用TRIzol法提取灰树花子实体总RNA,并用RNAeasy plant mini kit对提取的总RNA进行纯化,70℃变性2min后,NanoDrop ND-2000检测其浓度、琼脂糖凝胶电泳分析RNA 的完整性[6].检测合格的RNA 用于mRNA的富集及cDNA的合成.用Ultra RNA Library Prep Kit for Illumina进行文库构建,纯化后Agilent High Sensitivity DNA Kit检测文库插入片段大小,定量后Illumina HiSeqTM2000对建好的测序文库进行测序[7].
1.2.2 测序数据分析
使用Trinity(版本r20131110,默认参数)对RNA-seq的原始reads数据进行拼接,最短contig长度为200.对Trinity拼接结果使用Cap3进行进一步拼接获得Unigene[8].
1.2.3 Unigene的NR 数据库比对分析
利用Blast进行Unigene的NR 数据库物种分布比对分析[9],统计Blast结果中每个能比对上的物种所对应的Unigene数目,按该数目从高到低进行排序,选取数目较高的前10个物种,其他比对上的物种对应的Unigene数目相加作为others,没有比对上的物种的Unigene数目相加则是unmatched.
1.2.4 Unigene的GO 分类
根据NR 注释信息,对Unigene进行GO 注释[10](Blast2GO),得到每个Unigene的GO 注释.并对所有Unigene做GO 功能分类统计(WEGO),从细胞组成、分子功能及生物过程(biological process)三方面认识灰树花的基因功能分布特征.
1.2.5 Unigene的COG 功能注释
将Unigene和COG 数据库(http:∥www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi)比对分析,预测Unigene功能并对其分类统计,这有利于我们进一步了解灰树花各Unigene的生物学功能.
1.2.6 Unigene的代谢通路分析
使用http:∥www.genome.jp/tools/kaas/对Unigene进行KEGG 注释,以便于进一步研究灰树花基因在生物学上的复杂行为,系统分析其基因在细胞中的代谢通路及功能.
1.2.7 Unigene的SSR 信息分析
对拼接得到Unigene进行SSR 简单重复序列的查找.筛选标准:单核苷酸重复的次数在10次或10次以上,二核苷酸重复的次数在6次或6次以上,三至六核苷酸重复的次数在5次或5次以上.同时,也筛选中间被少数碱基(间隔小于100或等于100)打断的不完全重复的SSR.利用MISA(http:∥pgrc.ipkgatersleben.de/misa/)工具提供批量识别和定位简单重复序列(SSR).
2 结果与分析
2.1 RNA提取及Unigene的组装
灰树花子实体RNA 经过NanoDrop定量后,获得浓度为381.1ng/μL 的总RNA,260/280为2.12.完整性及28S∶18S(图1)均符合转录组测序质量要求,进入下一步实验.利用Trinity对RNA-seq的原始reads数据进行拼接,获得的contig进一步拼接获得Unigene.最终,我们获得63 137个Unigene,资料组总长度155 171 094nt,最长Unigene 20 996nt,最短Unigene 201nt,平均组装长度为2 457.689nt,(G+C)/(A+T+G+C)为0.522,N50为3 405nt,N90为1 390nt.

图1 灰树花子实体RNA 电泳检测Fig.1 The electrophoresis detection of maitake RNA

图2 Unigene长度分布统计Fig.2 Length distribution of maitake Unigene
从Unigene的长度分布来看,Unigene主要集中在1 500~10 000nt之间(图2).在2 000~3 000nt之间的Unigene数量最多为13 135个,占总数的20.8%;1 500~2 000nt之间Unigene数为8 979,占总数的14.2%;3 000~4 000nt之间Unigene数为7 429,占11.76%.
2.2 Unigene的NR 数据库比对分析
将Unigene序列和NR 数据库进行Blast(参数为1.0×10-5)比对分析,能比对上的Unigene个数为46 640 个,占总的Unigene数目的百分比为73.87%.按物种分布统计,能比对上的物种所对应的Unigene数目最多的是变色栓菌,比对上的Unigene数为14 593 个,占31.29%,其次为木质素降解菌,比对上的Unigene数为9 220个,占19.77%(表1).

表1 灰树花Unigene的NR 数据库比对分析Tab.1 Blast results of maitake Unigene via NR
2.3 Unigene的GO 分类
对Unigene进行GO 注释和GO 功能分类.最终(图3),在细胞组成(Cellular component)、分子功能(Molecular function)、生物过程(Biological process)3 个本体中,分别获得9,12,14个注释条目(Class)数.注释到的Unigene数量最多的是生物过程本体,注释到的Unigene数最少的是细胞成分本体.
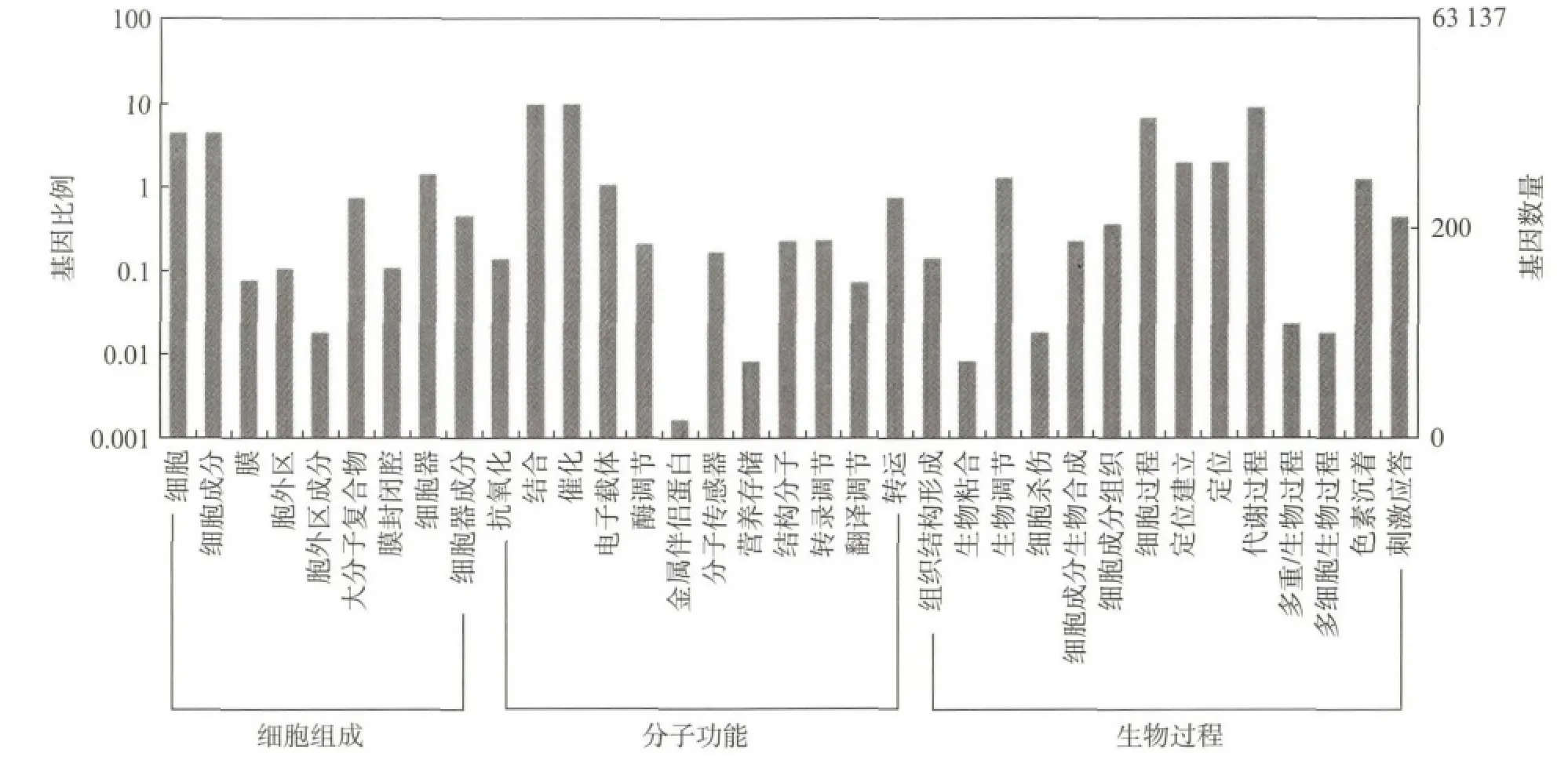
图3 灰树花Unigene的GO 分类Fig.3 Gene ontology classifications of maitake Unigene
2.4 Unigene的COG 分类
将所测物种的最佳蛋白序列提交到NCBI上(COG-4873PSSMs,E-value 0.01,Maximum number of hits 500),得到与Unigene编号相对应的COG 编号,统计COG 每个类别Unigene数目.从基因数量分布来看(图4),分类最多的是功能预测蛋白,其他较多的分类与基因的功能与糖转运与代谢、脂质转运与代谢、氨基酸转运和代谢、翻译后修饰、蛋白转换、分子伴侣有关.

图4 灰树花Unigene的COG 分类Fig.4 COG functional classifications of maitake Unigene
2.5 Unigene的代谢通路分析
KEGG 数据库(http:∥www.genome.jp/kegg/)可系统分析其基因在细胞中的代谢通路及功能.通过KEGG 注释,共有27 472 个Unigene被注释,被Unigene注释到的代谢通路有239个,注释最多的代谢通路与生化代谢、微生物代谢、次生代谢产物生物合成、嘌呤代谢、RNA 运输等有关(表2,注释到基因数前10位代谢通路).
2.6 Unigene的SSR 信息分析
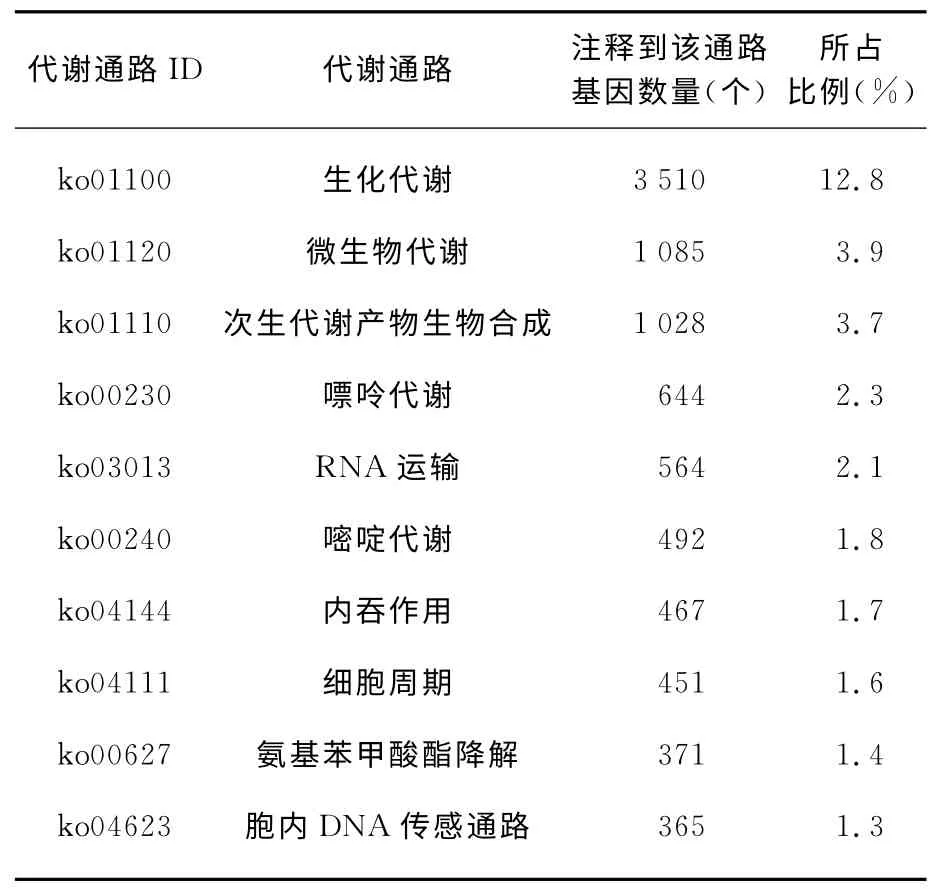
表2 KEGG 注释比例最多的前10位代谢通路Tab.2 The top 10pathways annotated by KEGG
从灰树花63 137个Unigene中查找到5 294个SSR位点,占Unigene总数的比例为8.38%(表3).SSR 存在较为丰富的类型,包括单核苷酸重复类型至六核苷酸重复类型均有表现(表4).其中,单核苷酸重复所占比例最高,达到63.4%,其次是三核苷酸重复,为24.44%,双核苷酸重复,比例为8.76%;比例最低的是六核苷酸重复,仅为0.11%,四核苷酸重复和五核苷酸重复基本相同,分别为1.81%和1.42%.在检出的SSR 中,出现频率最高的重复基元为A/T(占56.10%),其次为CCG/CGG(6.18%),AG/CT(3.29%),ACAGG/CCTGT(1.28%),AATG/ATTC(0.62%),AACAGC/CTGTTG(0.11%).上述SSR 特征分析,有助于开展灰树花及其同属物种的基因组差异分析、通用性标记开发和遗传图谱构建研究.

表3 灰树花的SSR 信息分析Tab.3 General statistics of maitake SSR search

表4 灰树花SSR 基序重复类型统计Tab.4 Statistics of repeat type of maitake SSR motif
3 讨论
对灰树花全基因组而言,其转录组序列不含内含子及其它非编码序列,能更高效的挖掘有用信息,在序列分析方面具有性价比高的优势.转录组研究可识别灰树花子实体总转录本的表达,从而了解灰树花子实体完整的基因表达谱,为灰树花具有生物功能的“蛋白质组”研究的必然纽带.基于灰树花总转录水平的研究是目前研究最广泛的调控研究方式[11].
本研究构建了第一个高质量灰树花cDNA 文库,首次采用了Illumina高通量测序技术对文库进行了测序,序列拼接后得到63 137个Unigene.将Unigene序列和NR 数据库进行Blast比对分析,能比对上的Unigene占总Unigene数的73.87%.COG 分类显示,最多一类基因是功能预测蛋白,其他较多基因功能与糖转运与代谢、脂质转运与代谢、氨基酸转运和代谢、翻译后修饰、蛋白转换、分子伴侣有关.该结果显示,利用高通量测序不仅可监测灰树花特定时间段的基因表达,更可大量挖掘其代谢过程中的重要基因.
根据KEGG 代谢通路数据库,对所得灰树花转录组的Unigene进行代谢通路注释和预测,共有27 472个Unigene被注释,被Unigene注释到的代谢通路有239个,该类基因参与了灰树花子实体体内的生化合成和次生产物代谢,研究该类基因,将为开展灰树花基因克隆、功能基因验证等分子手段提供生物信息学基础.
本次试验通过SSR 位点查找共发现5 294个SSR 位点,利用SSR 位点,筛选目的条带清晰、多态性好的引物,从而为分析灰树花群体遗传多样性、构建灰树花遗传连锁图谱、进行灰树花的分子育种奠定基础.
[1]周昌艳,吴爱忠,唐庆九,等.高分子量灰树花多糖GFLP的分离纯化及其对免疫细胞的作用[J].食用菌学报,2013,20(4):39-42.
[2]杨 海,耿传信,周学锋.灰树花活性多糖药理研究综述[J].中国执业药师,2012,9(3):30-33.
[3]张美彦,尚晓冬,郭 倩,等.人工控制条件下的灰树花菌株筛选[J].食用菌学报,2010,17(3):25-28.
[4]Yang F,Xu B,Li J J,et al.Transcriptome analysis of Termitomyces albuminosus reveals the biodegradation of lignocellulose[J].Acta Microbiologica Sinica,2012,52(4):466-477.
[5]陈士林,朱孝轩,陈晓辰,等.现代生物技术在人参属药用植物研究中的应用[J].中国中药杂志,2013,38(5):633-639.
[6]Tomas Maseka,Vaclav Vopalenskya,Petra Suchomelovab,et al.Denaturing RNA electrophoresis in TAE agarose gels[J].Analytical Biochemistry,2005,336(1):46-50.
[7]Shen R,Fan J B,Campbell D,et al.High-throughput SNP genotyping on universal bead arrays[J].Mutat Res,2005,573(1/2):70-82.
[8]Haas B J,Papanicolaou A,Yassour M,et al.De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis[J].Nature Protocols,2013,8(8):1494-1512.
[9]Quevillon E,Silventoinen V,Pillai S,et al.InterProScan:Protein domains identifier[J].Nucleic Acids Research,2005,33(2):116-120.
[10]Ye J,Fang L,Zheng H,et al.WEGO:A web tool for plotting GO annotations [J].Nucleic Acids Research,2006,34(2):293-297.
[11]Trapnell C,Williams B A,Pertea G,et al.Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation [J].Nature Biotechnology,2010,28(5):511-515.
猜你喜欢
井冈教育(2022年2期)2022-10-14 03:11:40
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
小品文选刊·印象大同(2022年11期)2022-06-10 21:13:01
动漫界·幼教365(大班)(2021年3期)2021-04-06 04:30:16
汉字汉语研究(2020年4期)2020-02-01 07:14:18
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
学生天地(2019年36期)2019-08-25 08:59:44
红领巾·萌芽(2019年4期)2019-05-17 21:53:07
食品工业科技(2014年13期)2014-03-11 18:16:46
