
Identification,synthesis and characterization of process related impurities of benidipine hydrochloride,stress-testing/stability studies and HPLC/UPLC method validations☆
2015-11-17 01:23:57EsenBellurAticiBekirKarl
Esen Bellur Atici,Bekir Karlığa
Deva Holding A.Ş.,Çerkezköy-2 Production Plant,Karaağaç Mh.Fatih Blv.No:26,Adres No:2278035833,Kapaklı,Tekirdağ,Turkey
Original Article
Identification,synthesis and characterization of process related impurities of benidipine hydrochloride,stress-testing/stability studies and HPLC/UPLC method validations☆
Esen Bellur Atici*,Bekir Karlığa
Deva Holding A.Ş.,Çerkezköy-2 Production Plant,Karaağaç Mh.Fatih Blv.No:26,Adres No:2278035833,Kapaklı,Tekirdağ,Turkey
A R T I C L E I N F O
Article history:
13 January 2015
Accepted 3 February 2015
Available online 13 February 2015
Benidipine
Impurities
Synthesis
Characterization
Validation
Stability
Benidipine hydrochloride,used as an antihypertensive agent and long-acting calcium antagonist,is synthesized for commercial use as a drug substance in highly pure form.During the synthetic process development studies of benidipine,process related impurities were detected.These impurities were identified,synthesized and characterized and mechanisms of their formation were discussed in detail. After all standardization procedures,they were used as reference standards for analytical studies.In addition,a separate HPLC method was developed and validated for detection of residual 1-benzylpiperidin-3-ol(Ben-2),which is used during benidipine synthesis and controlled as a potential process related impurity.As complementary of this work,stress-testing studies of benidipine were carried out under specified conditions and a stability-indicating UPLC assay method was developed,validated and used during stability studies of benidipine.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Benidipine hydrochloride,(±)-(4R*)-3-(R*)-1-benzylpiperidin-3-yl5-methyl1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate hydrochloride,is a racemic mixture of two isomers RR-(-)and SS-(+)and called α-benidipine hydrochloride(Fig.1).
Benidipine is a dihydropyridine vasoselective long acting calcium channel blocker developed in Japan[1].It blocks three different types of calcium channels(L,N and T)in the cell membrane by its unique mechanism of action.Benidipine has different unique chemical properties compared to the other calcium channel blocker agents[2],such as strong and long acting effect due to its high affinity and unique membrane binding sites,renal protective effect due to its triple(L,N and T)calcium channel blockage,and cardioprotective and vasoprotective effect due to its vascular selectivity and positive effect on nitric oxide production.As a solid,benidipine hydrochloride is stable to variations in heat and moisture,and is fairly stable to light exposure[3].
Several methods are reported in the literature[4]for synthesis of benidipine hydrochloride.Our synthesis method for benidipine hydrochloride is given in Scheme 1.During the synthesis and purification stages of benidipine hydrochloride,process related impurities were consistently observed in HPLC analyses.A few analytical methods have been reported in the literature for the determination of benidipine including studies related to its tablet and plasma concentrations[5-10].
As per the general guidelines recommended by the International Conference on Harmonisation(ICH)[11,12]to qualify the drug substance,the amount of acceptable level for a known and unknown related compound(impurity)should be less than 0.15% and 0.10%,respectively.In order to meet the stringent regulatory requirements,impurities should be identified and their amounts should be controlled carefully.In recent years,the impurity profile of a drug substance has become more important for marketing approval and this work is done as part of a drug development process[13-21].Of course,it is a challenging work for the development team to identify the impurities formed in very small quantities in a drug substance.Since mostly it is difficult to identify and control impurities within specified limits and additional purification steps may then be applied,it makes the process less competitive.Most of the impurities are not commercially available and they should be synthesized by the development team,which is not so easy since their syntheses are not known or not described in the literature.To the best of our knowledge,there is no report in the literature for the identification and synthesis of the impurities of benidipine.We believe that development of a drug substance is incomplete without the identification of animpurity profile involved in the process.Thus,in our study we explored the formation,identification,synthesis and characterization of impurities found in the synthesis of benidipine.This study will be of immense help for pharmaceutical development teams to understand the potential impurities in benidipine synthesis and thereby obtain the pure compound.
The impurities observed on HPLC analyses during the process development of benidipine(Fig.1)were identified by LC-MS.After thorough evaluation of the benidipine synthetic route(Scheme 1),all plausible side reactions were determined,and these reactions were carried out separately to synthesize the related impurities of benidipine hydrochloride.The synthesis and characterization of benidipine impurities were carried out and reported herein,together with the discussion on pathway of their formation.In addition,due to its highly polar property,a separate HPLC method was developed and validated for detection of residual 1-benzylpiperidin-3-ol(Ben-2),which was used during benidipine synthesis and controlled as a potential impurity.

Fig.1.Chemical structure of α-benidipine hydrochloride.

Scheme 1.Synthesis of benidipine hydrochloride.
The purpose of stability testing is to provide evidence on how the quality of a drug substance varies with time under the influence of a variety of environmental factors,such as temperature,humidity,and light,and to establish a retest period for the drug substanceandrecommendedstorageconditions[22,23].Ascomplementary of this work,stress-testing and stability studies of benidipine were carried out under specified conditions and a stability-indicating UPLC assay method was developed,validated and used during stress-testing and stability studies of benidipine. Unlike the known assay method for benidipine hydrochloride dependent on potentiometric titration[24]and not useful for assay analyses of stress-testing studies,our developed method is a stability-indicating,rapid,reliable,safe and clean method in terms of the chemicals used in both methods.
2.Experimental
2.1.Chemicals,reagents and samples
Benidipine hydrochloride samples were taken from commercial batches produced by Deva Holding A.Ş.(Tekirdag˘,Turkey),or synthesized in the R&D laboratories of Deva.Benidipine hydrochloride standard was supplied by a specialized team on standardization of reference standards for analytical use in Deva. Synthetic and analytical reagents and solvents were supplied from different chemical companies such as potassium dihydrogen phosphate,phosphoric acid,hydrochloric acid(37%),sodium hydroxide,hydrogen peroxide(30%),thionyl chloride,sulfuric acid(conc.)andsodiumhydrogencarbonatefromMerckKGaA(Darmstadt,Germany);tetrahydrofuran(THF),dichloromethane,acetonitrile,methanol,ethanol and acetone from J.T.Baker(Phillipsburg,USA);diethyl ether and ethyl acetate from Lab-Scan(Gliwice,Poland);chromium(VI)oxide(CrO3)from Sigma-Aldrich(St.Louis,MO,USA);potassium hydroxide from Acros Organics(Geel,Belgium);dimethyl formamide from Akkim (Yalova,Turkey);anhydrous sodium sulfate from Sodas Sodyum Sanayi A.S.(Izmir,Turkey),and sodium chloride from Emekcioglu Tuz A.S.(Ankara,Turkey).Deionized water was prepared using MilliQ plus purification system (Millipore,Bedford,MA,USA).Deuterated solvents(dimethylsulfoxide-d6 and deuterated chloroform)were purchased from Merck KGaA(Darmstadt,Germany).
2.2.Liquid chromatography(HPLC and UPLC)
Chromatographic separations were performed on HPLC system with Waters Alliance 2695 separation module equipped with a Waters 996 photodiode array detector and an Empower-pro data handling system(Waters Corporation,Milford,MA,USA).
The analysis of all compounds excluding Ben-2 was carried out on C18(100 mm×4.6 mm,3 μm)column(Hypersil BDS)at 25°C with phosphate-buffer/methanol/THF(65/27/8)mixture flowing at a rate of 0.75 mL/min(it was adjusted during the analysis so that the retention time of benidipine was about 20 min)during 60 min.Buffer solution was prepared by dissolving 6.8 g of potassium dihydrogen phosphate in 1000 mL water and pH was adjusted to 3.0±0.05 with 30%phosphoric acid.The injection volume and detection wavelength were fixed at 10 μL and 237 nm,respectively.Test solution was prepared by dissolving 20.0 mg of sample in 100.0 mL diluent(0.2 mg/mL)and methanol/water(50/ 50)mixture or methanol was used as diluent.
The analysis for determination of Ben-2 impurity in benidipine was carried out on C1(250 mm×4.6 mm,5 μm)column(ACE C1)at 25°C with mobile phase flowing at a rate of 1.0 mL/min for 27 min.The separation was employed using gradient elution with phosphate-buffer(mobile phase A)and acetonitrile(mobile phase B)based on the following program:time(min)/%of mobile phase B:0/10,6/10,15/60,20/60,21/10,27/10 and the dwell volume of the equipment was 650 μL.Buffer solution was prepared by dissolving 6.8 g of potassium dihydrogen phosphate in 1000 mL water and pH was adjusted to 6.0±0.05 with 10%potassium hydroxide.The injection volume and detection wavelength were fixed at 10 μL and 210 nm,respectively.Test solution(1.0 mg/mL)was prepared by dissolving 50.0 mg of benidipine hydrochloride in 50.0 mL methanol/water(50/50)mixture and the concentration of Ben-2 reference solution was 1.0 μg/mL.
Assay studies were performed on Waters Acquity UPLC system equipped with a Waters TUV detector and an Empower-pro data handling system (Waters Corporation,Milford,MA,USA).The analysis was carried out on BEH Shield RP18(100 mm×2.1 mm,1.7 μm)column(Waters Acquity)with 1 μL injection volume at a wavelength of 237 nm and with phosphate-buffer/acetonitrile(60/ 40)mixture flowing at a rate of 0.3 mL/min during 5 min.Column and sample temperatures were 30°C and 25°C,respectively. Buffer solution was prepared by dissolving 6.8 g of potassium dihydrogen phosphate in 1000 mL water and pH was adjusted to 3.0±0.05 with 30%phosphoric acid.Methanol/water(50/50)was used as diluent.Test and standard solutions were prepared by dissolving 20.0 mg of sample and reference standard,respectively,in 100 mL of diluent(0.2 mg/mL).
2.3.Mass spectrometry
The mass spectra were recorded on Waters LC-MS ZQ 2000/ 4000 system (Waters Corporation,Milford,MA,USA).The fragmentation profile of the samples was established by carrying out MS studies in positive or negative electrospray ionization(ESI)mode.For LC-MS identification of the impurities,the HPLC method given above was used.The samples of the synthesized compounds were directly infused using a syringe at a concentration of 1 mg/mL in methanol.
2.4.NMR spectroscopy
1H,13C and DEPT NMR experiments were performed on a 300 MHz NMR Spectrometer(Varian NMR Instruments,Oxford,UK;later acquired by Agilent Technologies,Santa Clara,CA,USA)using deuterated dimethylsulfoxide(DMSO-d6)and deuterated chloroform (CDCl3)as solvent and tetramethylsilane(TMS)as an internal standard.
2.5.FT-IR spectroscopy
Samples were measured as neat by ATR(Attenuated Total Reflectance)on Shimadzu FTIR Spectrometer IR Prestige-21(Shimadzu Corporation,Kyoto,Japan)in the range of 600-4000 cm-1with 20 scans and 2 cm-1resolution.
2.6.Differential scanning calorimetry(DSC)and melting point
determination
DSC measurements were carried out using a Shimadzu DSC-60 instrument(Shimadzu Corporation,Kyoto,Japan).The instrument was calibrated for temperature and heat flow using indium and zinc standards.The samples(2-3 mg)were placed in sealed aluminum pans under nitrogen purge at a flow rate of 30 mL/min.The samples were heated from 100°C to 220 or 300°C depending on the melting range of the compound at a heating rate of 10°C/min.Data acquisition and analysis were performed using the Shimadzu TA-60WS software. Start and end points for the integration of the thermal peak were identified by visual inspection.The amounts of samples were weighed in a Sartorius analytical balance model ME235S(Sartorius AG,Gottingen,Germany),with a resolution of 0.01 mg.Melting points were determined on an Electrothermal 9100 digital melting point instrument(Thermo Fisher Scientific,Essex,UK).
2.7.Synthesis of benidipine and its impurities
2.7.1.Synthesis of α-and β-benidipine hydrochloride(Scheme 1)
2.7.1.1.Synthesis of α-benidipine hydrochloride.1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid3-methyl ester(Ben-1)(1.00 equiv.)was suspended in dichloromethane and dimethyl formamide(4:1)mixture and the suspension was cooled to 0-5°C.Thionyl chloride(1.20 equiv.)was added to this mixture and stirred for 1 h.Then,1-benzyl-3-piperidinol(Ben-2)(1.10 equiv.)was added into the reaction mixture and stirred for 2 h at 0-5°C and 1 h at 25°C.The reaction was quenched by addition of water.Organic phase was separated,washed with brine,dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to dryness under vacuum and the residue was crystallized in ethanol and acetone mixture.The crude product was filtered and dried under vacuum.After several recrystallization processes applied using ethanol and acetone,pharmaceutical grade α-benidipine hydrochloride was obtained as yellowish crystalline powder with overall 45%yield.
2.7.1.2.Isolation of β-benidipine hydrochloride(β-isomer impurity). All crystallization filtrates were combined during the benidipine hydrochloride synthesis(Section 2.7.1.1)and concentrated to a volume until β-isomer started to crystallize.The suspension was stirred for 2 days at room temperature and filtered to obtain βbenidipine hydrochloride as yellowish crystalline powder(36%).
2.7.2.Synthesis of Ben-ox impurity(3-(1-benzylpiperidin-3-yl)
5-methyl 2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate hydrochloride)(Scheme 2)
To the suspension of benidipine hydrochloride(1.00 equiv.)in acetone,Jones reagent[25](prepared by dissolving 81 g of CrO3in 69 mL of concentrated sulfuric acid followed by cautious dilution with water to 300 mL)was added at 0-5°C and the suspension was completely dissolved.After completion of the reaction(control by HPLC)in approximately 1 h,the reaction was neutralized by addition of saturated aqueous sodium hydrogen carbonate solution and the reaction mixture was extracted with ethyl acetate. Organic extract was washed with brine,dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to dryness under vacuum.The residue was dissolved in diethyl ether and washed with saturated aqueous sodium hydrogen carbonate solution.The organic phase was dried over anhydrous sodium sulfate and filtered,and the filtrate was concentrated to dryness under vacuum.The product Ben-ox base“3-(1-benzylpiperidin-3-yl)5-methyl2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate”was obtained as dark yellowish/brownish viscous oil(99.6%).The product was then converted to its hydrochloride salt by hydrochloric acid treatment in dichloromethane,dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to dryness under vacuum to obtain the product Ben-ox“3-(1-benzylpiperidin-3-yl)5-methyl 2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate hydrochloride”as slightly yellowish foamy powder(97.4%).
2.7.3.Synthesis of Ben-bis impurity(bis(1-benzylpiperidin-3-yl)1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate dihydrochloride)(Scheme 3)
2.7.3.1.Synthesis of Ben-1 acid“1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylic acid”.Suspension of Ben-1“5-(methoxycarbonyl)-1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3-carboxylic acid”(1.00 equiv.)in water was cooled to 0-5°C and potassium hydroxide pellets(15.0 equiv.)were added slowly.After the addition,the mixture was allowed to warm to ambient temperature and stirred until completion of the ester hydrolysis,which was monitored by HPLC and took around 6-7 days.The reaction mixture was then filtered and the filtrate was washed with diethyl ether,ethyl acetate,and dichloromethane,respectively.Aqueous phase was cooled to 0-5°C and neutralized by addition of concentrated hydrochloric acid.The formed suspension was stirred at this temperature for 10 min,filtered,washed with water and dried under vacuum to obtain Ben-1 acid“1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-di carboxylic acid”as beige powder(62%).
2.7.3.2.Synthesis of Ben-bis impurity.Ben-1 acid“1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylic acid”(1.00 equiv.)was suspended in dichloromethane and dimethyl formamide(4:1)mixture and the suspension was cooled to 0-5°C. Thionyl chloride(2.40 equiv.)was added to this mixture and stirred for 1 h.Then,Ben-2“1-benzyl-3-piperidinol”(2.20 equiv.)was added into the reaction mixture and stirred for 2 h at 0-5°C and for 1 h at 25°C.The reaction was quenched by addition of water. Organic phase was separated,washed with brine,dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to dryness under vacuum and the residue was crystallized in ethanol and acetone mixture.The product was filtered and dried under vacuum to obtain Ben-bis“bis(1-benzylpiperidin-3-yl)1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate dihydrochloride”as slightly yellowish powder(20%).
2.7.4.Synthesis of Ben-desbenzyl impurity(3-methyl 5-piperidin-3-yl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate hydrochloride)(Scheme 4)
Ben-1“1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 3-methyl ester”(1.00 equiv.)was suspended in dichloromethane and dimethyl formamide(4:1)mixture and the suspension was cooled to 0-5°C.Thionyl chloride(1.20 equiv.)was added to this mixture and stirred for 1 h.Then,piperidin-3-ol(1.10 equiv.)was added into the reaction mixture and stirred for 2 h at 0-5°C and for 1 h at 25°C.The reaction was quenched by addition of water.The organic phase was separated,washed with brine,dried over anhydrous sodium sulfate and filtered.The filtrate was concentrated to dryness under vacuum and the residue was crystallized in ethanol and acetone mixture.The product was filtered and dried under vacuum to obtain Ben-desbenzyl“3-methyl 5-piperidin-3-yl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate hydrochloride”as a slightly yellowish powder(25%).
2.8.Stress-testing and stability studies
Benidipine was subjected to stress-testing under the following conditions:neutral hydrolysis(stirring in water at 100°C for 4 h),alkaline hydrolysis(stirring in 1.0 M NaOH solution at 25°C for 4 h),acidic hydrolysis(stirring in 1.0 M HCl solution at 25°C for 4 h),oxidative degradation(stirring in 3%H2O2solution at 25°C for 4 h);thermal(standing at 100°C for 3 days),photolytic degradation under UV-light(standing under near ultraviolet lamp(254-400 nm,NLT 200 W h/m2)at 25°C for 3 days)and day-light(standing under cool white fluorescent lamp(1.2 million lx h)at 25°C for 3 days).The degraded samples were neutralized(for acidic and basic hydrolysis)and diluted to obtain 0.2 mg/mL solutions.Then,the samples were analyzed to determine the formed degradation impurities and benidipine amount remained.For each study,the corresponding blank solutions were prepared.Stability studies were conducted under accelerated(40±2°C,75%±5% humidity)and long-term (25±2°C,60%±5%humidity)conditions,and samples were analyzed according to the related substances and assay methods reported herein.
3.Results and discussion
3.1.Detection and identification of impurities
A typical analytical HPLC chromatogram of a production batch of benidipine bulk drug spiked with impurities is shown in Fig.2. The impurities(marked as Ben-desbenzyl,Ben-bis,Ben-ox,β-isomer and Ben-1)were detected in the crude sample of benidipine during process development studies and their identification was performed by LC-MS.They were subsequently synthesized to obtain sufficient quantities for full characterization and further analytical studies.All synthesized impurities were co-injected with benidipine to confirm the identity of the impurities based on retention time matching(Fig.2).Also,they were separately spiked inside of the crude benidipine samples where they appeared as an impurity on HPLC chromatograms.All impurities were well resolved from benidipine and each other.Retention time(RT,min),relative retention times(RRT)of the impurities with respect to benidipine,purity,DSC peak or melting point,IR and mass data,and structures are shown in Table 1 and Fig.3,respectively.1H and13C NMR assignments for benidipine and its impurities are shown in Tables 2 and 3,respectively.
3.2.Structure elucidation and formation of impurities
The impurity at RT 26.96 min was very rarely observed in the crude product during laboratory trials in the range of 0.05%-0.50% and never observed in commercial productions after optimization of the reaction and recrystallization steps of the process.The ESI mass spectrum exhibited a molecular ion at m/z 331 in negative ion mode,indicating molecular weight of 332,which was 173 amu lower than that of benidipine.According to the mass data,this impurity was identified as one of the benidipine intermediates called“5-(methoxycarbonyl)-1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3-carboxylic acid”and named as Ben-1 impurity(Fig.3,Scheme 1).Ben-1 is a process related impurity and may be found in the crude benidipine samples in case of incomplete reaction.High-purity Ben-1 was prepared,fully characterized and standardized for further analytical studies.
The impurity at RT 23.12 min was observed in crude benidipine during process development studies and commercial productions. The ESI mass spectrum exhibited a molecular ion at m/z 506 in positive ion mode,indicating molecular weight of 505,which is identical to that of benidipine.Its UV spectrum was also similar to the benidipine spectrum and it was the main impurity observed in the crude product samples,almost 30%-40%before the process optimization.As we know,benidipine has two stereogenic centers and these stereocenters cause four enantiomers and two diastereomers which are called α-benidipine(RR and SS racemic mixture)and β-benidipine(RS and SR racemic mixture)(Fig.3,Scheme 1). According to the benidipine synthesis given in Scheme 1,reaction resulted in the formation of both α-and β-benidipine almost in a one-to-one ratio.α-Benidipine is the diastereomer that is used as an active pharmaceutical ingredient and observed at 20.38 min on HPLC chromatogram given in Fig.2.Due to the solubility differences between α-and β-isomers,β-isomer was successfully removed by optimized crystallization processes;even crude product contained a small amount of this side product just after one crystallization process following the reaction.Under the light of all these information and spectral data,it was clarified that the impurity at RT 23.12 min was β-isomer of benidipine.This side product was isolated from crystallization filtrates of benidipine,fully characterized and standardized for analytical use.
The ESI mass spectrum exhibited a molecular ion at m/z 504 in positive ion mode,indicating molecular weight of 503,which was 2 amu lower than that of benidipine.This difference could be explained by oxidation of the 1,4-dihydropyridine moiety of benidipine to pyridine,which is commonly seen on the other 1,4-dihydropyridines used as an API in the pharmaceutical industry[26]. The impurity observed at RT 15.89 min was identified as oxidation impurity“3-(1-benzylpiperidin-3-yl)5-methyl 2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate”and named as Ben-ox impurity(Fig.3,Scheme 2),which was very rarely found in the crude product during process development studies(<0.05%),never observed in commercial productions and observed in the product during stability studies(0.05%-0.10%).This process related impurity was synthesized from benidipine by aromatization of the 1,4-dihydropyridine ring to pyridine,fully characterized and standardized for analytical use.Compared with benidipine NMR spectra,1H NMR spectrum(DMSO-d6)of Ben-ox impurity showed no signals for positions 1 and 4 and13C NMR and DEPT spectra showed signal at 137.7 ppm(C)for position 4,which was 39.3 ppm(CH)for benidipine.All spectral data confirmed the structure of Ben-ox impurity.

Fig.2.HPLC analysis of benidipine spiked with impurities.
The ESI mass spectrum of the impurity observed at RT 8.55 min exhibited a molecular ion at m/z 665 in positive ion mode,indicating molecular weight of 664,which was higher by 159 amu than that of benidipine.This difference could be explained by loss of methyl group and addition of a second 1-benzylpiperidinyl moiety to form “bis(1-benzylpiperidin-3-yl)1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate”(Fig.3).This impurity was named as Ben-bis and determined in all benidipinebatches ranging from 0.05%to 0.30%.The amount of Ben-bis impurity was dependent on the content of a Ben-1 impurity called Ben-1 acid“1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylic acid”.Formation of Ben-1 acid as a by-product during production of Ben-1 intermediate by alkaline ester hydrolysis of“dimethyl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate”was expected(Scheme 1).According to our synthetic and analytical studies,it was confirmed that Ben-1 contains this dicarboxylic acid impurity in the range of 0.15%-0.55%.Ben-1 acid impurity present in Ben-1 gave reaction with“1-benzylpiperidin-3-ol(Ben-2)”from both sites and formed the Benbis impurity in benidipine.Ben-1 acid was then prepared by ester hydrolysis of Ben-1,and reacted with Ben-2 under similar conditions used for synthesis of benidipine to obtain the Ben-bis impurity(Scheme 3).Ben-1 acid and Ben-bis impurities were then characterized and standardized for analytical use.Compared with benidipine NMR spectra,1H NMR spectrum(DMSO-d6)of Ben-bis impurity showed doubled signals for the 1-benzylpiperidinyl moiety and missed methoxy peak.13C NMR and DEPT spectra showed no methoxy peak,a single peak for both methyl groups(positions 7 and 8)located on 1,4-dihydropyridine ring,and also a single peak for carboxyl groups(positions 9 and 10)due to thesymmetric structure of the molecule.All spectral data confirmed the structure of Ben-bis impurity,which was observed in all benidipine batches during process development studies and commercial productions.Production process was especially optimized to eliminate this impurity and its amount was successfully reduced under 0.10%in the final product.
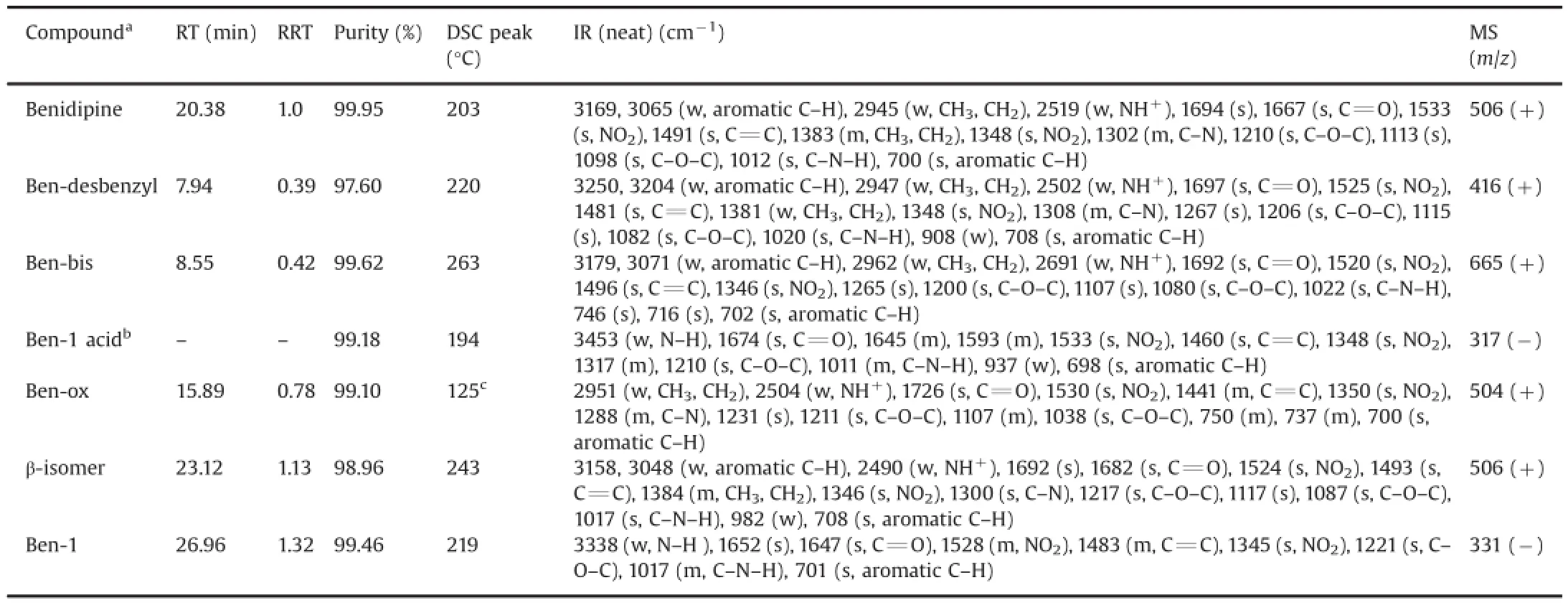
Table 1Retention time,relative retention time(RRT),purity,DSC peak or melting point,FTIR and mass spectral data of benidipine and its impurities.

Fig.3.Structures of benidipine and its impurities.
The ESI mass spectrum of the impurity observed at RT 7.94 min exhibited a molecular ion at m/z 416 in positive ion mode,indicating molecular weight of 415,which was 89 amu lower than that of benidipine.This difference could be explained by loss of benzyl group located on 1-benzylpiperidinyl moiety.This structure corresponds to the molecule called“3-methyl 5-piperidin-3-yl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarbox ylate”which was named as Ben-desbenzyl impurity(Fig.3).This impurity was formed by reaction of an impurity carried from previous steps of the synthesis,as in the case of formation of Benbis impurity.The possible pathway for formation of Ben-desbenzyl impurity could be explained by reaction of Ben-1 with“piperidin-3-ol”impurity present in Ben-2.“Piperidin-3-ol”impurity was formed during synthesis of Ben-2 by sodium borohydride reduction of the residual starting material“piperidin-3-one”present in“1-benzylpiperidin-3-one”.“Piperidin-3-ol”reacted with Ben-1 under similar conditions used for synthesis of benidipine to obtain the Ben-desbenzyl impurity(Scheme 4),which was then characterized and standardized for analytical use.Compared with benidipine NMR spectra,benzyl group signals were missed on both1H NMR and13C NMR spectra(DMSO-d6)of Ben-desbenzyl impurity.Also,higher amine(position 20)signal was found on1H NMR spectrum.All spectral data confirmed the structure of Bendesbenzyl impurity,which was observed in few benidipine batches during process development studies,never observed after optimization of the process conditions and during commercial productions.
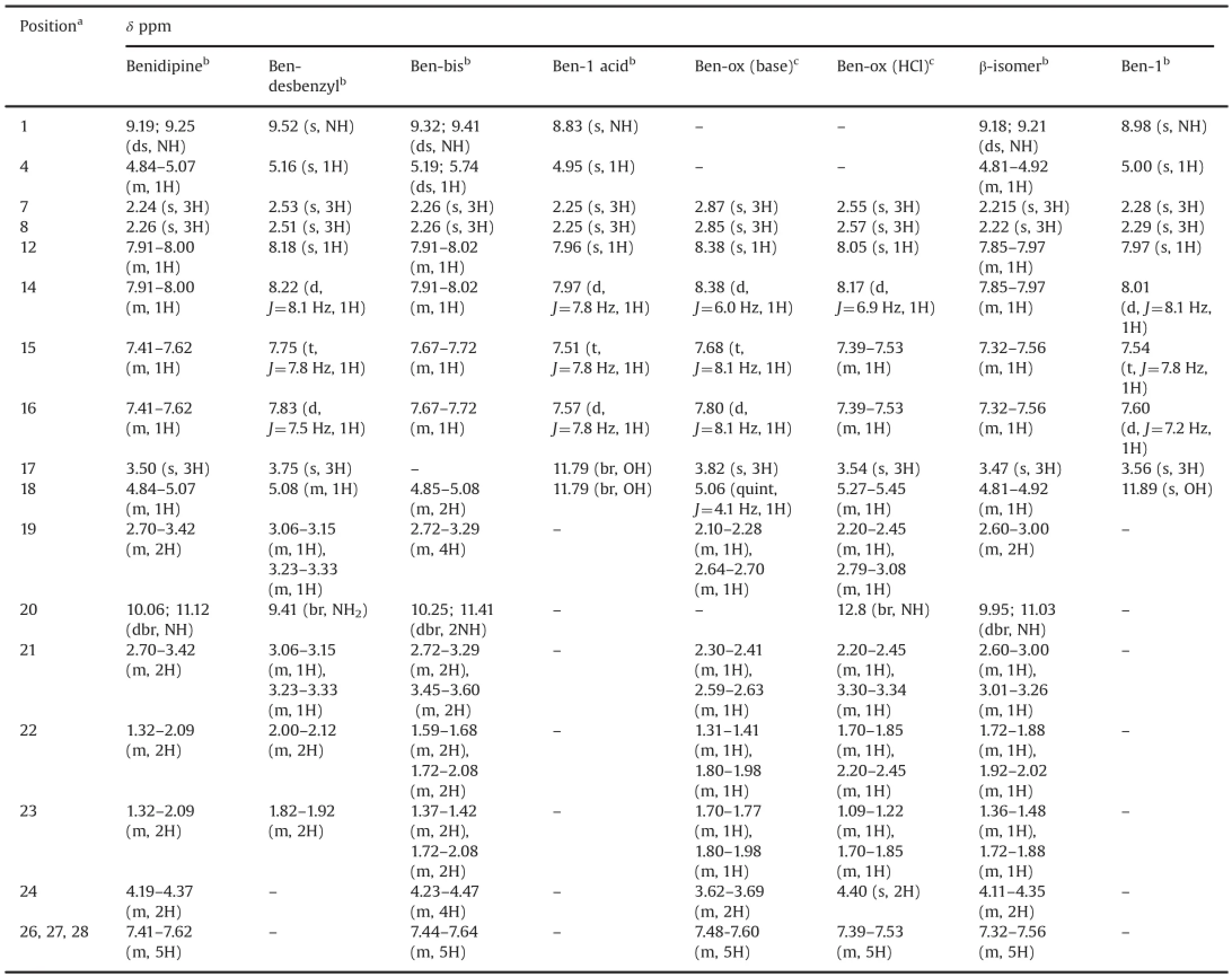
Table 21H NMR assignments for benidipine and its impurities.
3.3.Validation of HPLC method for determination of Ben-2
The method was validated according to ICH Q2(R1)guideline[27]and results of the validation study are given below.
3.3.1.System suitability
The system suitability was conducted throughout the validation study by using 1 μg/mL reference solution(Ben-2)and evaluated by making six replicate injections.The system was deemed to be suitable for use as the relative standard deviation(RSD)of the areas was below 5.0%.The average of the areas was found as 22089 with standard deviation(SD)87 and RSD was calculated as 0.4%,confirming the injection repeatability of the developed method.
3.3.2.Specificity and stability
System suitability test was conducted and RSD value was found to be 0.6%.The following injections were done for specificity test: one diluent,two test solutions(1 mg/mL benidipine hydrochloride),and two test solutions spiked with Ben-2 at the specification limit(0.10%);no overlapping peak was observed on diluent chromatogram at the retention time of Ben-2.Thus,the method was found specific for determination of Ben-2 in benidipine.Stability of solutions was evaluated by injection of two reference solutions and two test solutions spiked with Ben-2 at the specification limit freshly and then each 12 h for two days.Samples were accepted as stable for a time period during which the difference between fresh and different period results was below0.05%.For both solutions this difference did not exceed 0.01%with an RSD value of 1.3%and the samples were found stable up to 48 h. 3.3.3.Sensitivity(LOD and LOQ)and LOQ precision
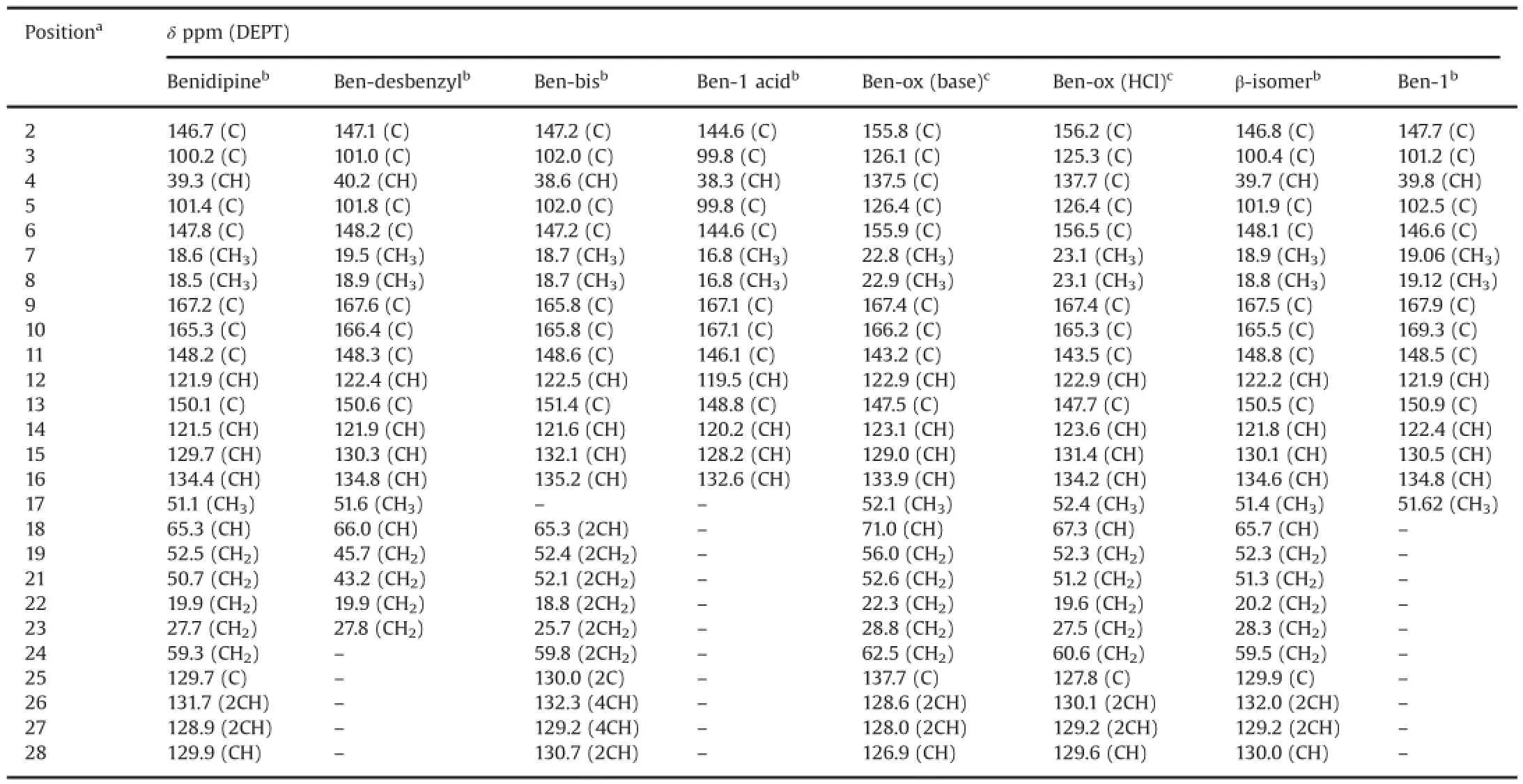
Table 313C chemical shifts in benidipine and its impurities.
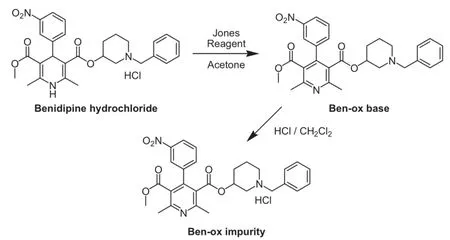
Scheme 2.Synthesis of Ben-ox impurity.
The LOD and LOQ for Ben-2 impurity were estimated at a signal-to-noise ratio of 3:1 and 10:1,respectively,by injecting six replicate reference solutions with 1 μg/mL concentration,and calculated according to the following equations:LOD=(3/SN)×C;LOQ=(10/SN)×C,where SN is signal-to-noise and C is concentration of the reference solution.LOD and LOQ were found to be 0.003%(0.03 μg/mL)and 0.01%(0.09 μg/mL),respectively.RSD value was found to be 0.4%(<5.0%)and LOQ was below the reporting level 0.05%and met the validation criterion.Precision at the LOQ level was carried out by injecting six replicates of standard solution of Ben-2 at LOQ concentration.Average of the areas was found as 2179 with standard deviation of 35,and RSD value of 1.6%,which was below the acceptance limit of 10.0%.Chromatograms of diluent,test solution spiked with Ben-2 at specification limit(0.10%),LOD(Ben-2 conc.0.03 μg/mL(0.003%))and LOQ(Ben-2 conc.0.09 μg/mL(0.01%))solutions are given in Fig.4.
3.3.4.Linearity,range,accuracy,precision and robustness

Scheme 4.Synthesis of Ben-desbenzyl impurity.
System suitability test was conducted and the RSD value was found to be 1.1%.Linearity test solutions of Ben-2 were prepared at five concentration levels ranging from LOQ to 150%of the specification level(0.10%)and each sample was injected three times. The peak area versus concentration data was analyzed with least squares linear regression.Correlation coefficient(r)and y-intercept ratio to the response of target concentration were found to be 0.99996(≥0.99)and 3.0%(≤5.0%),respectively,indicating the good linearity of the method.Solutions at level 1(LOQ)and level 5(150%of specification limit)were injected six times,and RSD value of areas was calculated for range study,and found to be 1.6%and 0.4%,respectively,below the acceptance limit of 5.0%.The accuracy of the method was evaluated on test solutions spiked with Ben-2 in triplicate at three concentration levels of 0.49 μg/mL,0.99 μg/mLand 1.48 μg/mL(50%,100%and 150%of the specification limit,respectively).Good-to-excellentrecoveriesofBen-2(99.32-100.45%)at each level were achieved within the limit range of 90.0%-110.0%(Table 4).Results for RSD and RSD of recoveries were found below 5.0%,namely 0.4%and 3.2%,respectively.The precision of the method was investigated by injecting two individual test solutions of benidipine hydrochloride(1 mg/mL)and six individual test solutions spiked with Ben-2 at the specification limit(0.10%).The same procedure was applied for the inter-day precision by a different analyst using different batch column and different instrument located within the same laboratory.The recoveries of Ben-2 for the intra-day and inter-day precision studies were obtained in the range of 97.98%-101.01%and 96.26%-100.93%,respectively.Results obtained for RSD of recoveries(1.12% and 2.28%)were below 5.0%in both studies(Table 4).Difference of the mean recoveries between two studies for intra-day and interday precision was found to be 0.7%and the method was found to be sufficiently precise since no significant variation in the found concentrations was observed on any day.Robustness of the method was tested by changing the flow rate by 10%(1.0±0.1 mL/ min),buffer pH by±0.2 units(6.0±0.2),and the column temperature by 5°C(25±5°C).In all of the above varied chromatographic conditions,RSD was found below 1.6%,and no difference was found between the normal and altered conditions,indicating the robustness of the method.
Requirements for each stage of the validation study were fulfilled.Thus,our developed method was precise,linear,accurate,sensitive,selective and robust,and could be used for determination of Ben-2 impurity in benidipine drug substance.This method was used to control residual Ben-2 as a potential impurity and to date has not been detected in any of the manufactured benidipine batches.
3.4.Validation of UPLC assay method
The known assay method for benidipine hydrochloride depends on the potentiometric titration,and is performed by addition of formic acid and acetic anhydride,respectively,into the sample solution and titration of the resulting mixture with perchloric acid[24].Unfortunately,this method is not stability-indicating and selective to benidipine itself,since it depends on titration of the amine group and all amine containing molecules are titrated including the impurities.Thus,it cannot be used for stress and stability testing where it is necessary to reach the mass balance.Due to the necessity of a new stability-indicating assay method for benidipine,we developed a method and validated it according to ICH Q2(R1)guideline[27].
3.4.1.System suitability
The system suitability was conducted throughout the assay validation study by using standard benidipine hydrochloride solution with a concentration of 0.2 mg/mL and evaluated by making six replicate injections.The system was accepted to be suitable for use as the relative standard deviation of the areas was below 0.85% and the symmetry factor below 2.0.Average of the areas was found as 1,638,857 with standard deviation(SD)2326 and RSD was calculated as 0.14%,confirming the system suitability of the developed method.
3.4.2.Specificity and stability
Specificity test was carried out by injections of three blank solutions and two test solutions and no peak was observed on diluent chromatogram at the retention time of benidipine.Stability of the test solution was evaluated by injection of two different test solutions freshly and then each 12 h for two days.Samples were accepted as stable for a time period during which the difference between fresh and different period results is below 2.0%;all assay results should be found in the range 98.0%-102.0%(on a dry basis).For both solutions this difference did not exceed 0.19% and all assay results were found in the range 98.3%-101.6%;thus the test solution was found stable up to 48 h.
3.4.3.Linearity,range,accuracy,precision and robustness
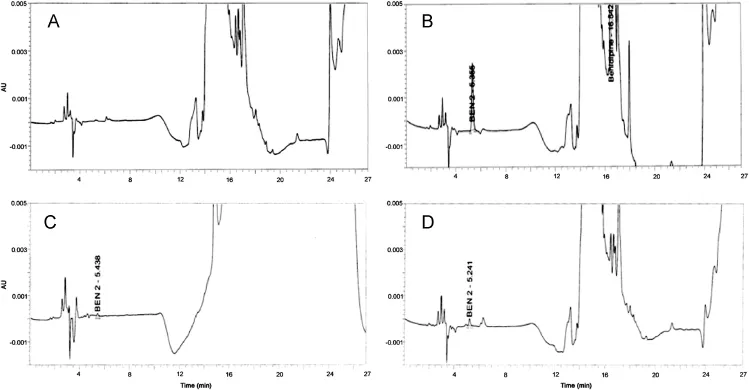
Fig.4.Chromatograms of HPLC method validation for determination of Ben-2 in benidipine:(A)blank solution,(B)test solution spiked with Ben-2 at specification limit(0.10%),(C)LOD solution(Ben-2 conc.0.03 μg/mL(0.003%)),and(D)LOQ solution(Ben-2 conc.0.09 μg/mL(0.01%)).
Linearity test solutions of benidipine hydrochloride were prepared at five concentration levels ranging from 80%to 120%of the target concentration(0.2 mg/mL)and each sample was injectedthree times.The peak area versus concentration data was analyzed with least squares linear regression.Correlation coefficient(r)and y-intercept ratio to the response of target concentration were found as 0.99998(≥0.999)and 1.9%(≤2.0%),respectively,indicating the good linearity of the method.Solutions at level 1(0.16 mg/mL)and level 5(0.24 mg/mL)were injected six times and RSD of areas was calculated for range study,and found below the acceptance limit of 0.85%,namely 0.18%and 0.10%,respectively. The accuracy of the method was evaluated on test solutions prepared in triplicate at three concentration levels(80%,100%and 120%of the target concentration)and each solution was injected twice.Good-to-excellent recoveries(100.2%-101.4%)at each level were achieved within the limit range of 98.0%-102.0%and RSD of recoveries was found to be 0.36%(Table 5).
The method precision of an analytical procedure expresses the closeness of agreement between single results of the method applied repeatedly by the same analyst,using the homogeneous sample,the same instrumentation and the same reagents.The precision of the method was investigated by injecting six individual test solutions.The inter-day precision was conducted according to the same procedure on a different day by a different analyst using different batch columns and different instruments located in the same laboratory.The assay results for intra-day and inter-day precision studies were obtained in the range of 98.6%-101.2%and 99.0%-100.3%(on dry basis),respectively.RSD values of the results were found to be 0.98%and 0.44%(limit:1.34%),respectively.Difference between the mean of results obtained for intra-day and inter-day precision(0.8%)was below the acceptance limit of 2.0%as recommended by the ICH guideline,indicating that the developed method was found to be precise.The robustness of an analytical method is a measure of the effect of variable conditions on analytical results in the process of analysis.For the determination of the method's robustness,flow rate,mobile phase composition,pH of the buffer and column temperature were varied within a realistic range and the effect of variations on analytical procedure was examined and reported.Robustness of the methodwastestedbychangingtheflowrateby10%(0.30±0.03 mL/min),buffer pH by±0.1 units(3.0±0.1),column temperature by± 5°C(30±5°C),and the mobile phase acetonitrile composition by±2%(40%±2%).In all of the above varied chromatographic conditions,RSD and symmetry factors did not exceed the limit values of 0.85%and 2.0,respectively,and were found below 0.64%and 1.5,respectively.Assay results were obtained in the range of 99.4%-100.6%(on dry basis),and difference of the mean assay results between the normal and altered conditions was found below 1.0%(limit:2.0%),indicating the robustness of the method.
The results showed that our developed assay method is specific,precise,accurate and robust,and it could be used for assay analyses of benidipine manufacturing batches.The known potentiometric titration method was also useful and successfully used for assay analyses of benidipine batches parallel to the developed UPLC method.But,unfortunately titration method was not stability-indicating and could not be used for assay analyses of benidipine during stress-testing and stability studies,where UPLC method was applied successfully.Compared with the potentiometric method the UPLC method is faster(5 min run time),safer and cleaner in terms of the chemicals used.The potentiometric method is also problematic due to the use of acetic anhydride which is in the controlled chemicals list of many countries and not easy to import,and needs to be followed very carefully.The UPLC assay method is also more reliable than potentiometric titration and could be used for assay analyses of benidipine drug products(tablets).
3.5.Stress-testing and stability studies
Stress-testing helps determine the intrinsic stability of the molecule by establishing degradation pathways in order to identify the likely degradation products and to validate the stability-indicating power of the analytical methods used.The nature of the stress-testing depends on the individual drug substance and the type of drug product involved.In this study,degradation products of benidipine formed under the influence of stress conditions were determined and the degradation pathway was predicted.According to the results given in Fig.5 and Table 6,benidipine was not affected from light and it was photo-stable;it was oxidized and formed Ben-ox impurity under neutral and acidic hydrolysis and thermal conditions;the most serious degradation was observed under oxidation condition where a new degradation product was detected at around 29 min(RRT 1.43)besides the Ben-ox impurity.Peak purity test results obtained from a PDA detector confirmed that the benidipine peak was homogeneous and pure in all of the analyzed stress samples,and the mass balance results were in the range of 99.9%-100.2%.This study helped us during selection of the packaging material and storage conditions of the drug substance and also formulation studies of the drug product.Benidipine was found stable after 6 months'storage at accelerated stability testing conditions.Also,benidipine stayed stable after two years of storage at normal conditions.

Table 4Accuracy and precision studies of the HPLC method for determination of Ben-2 in benidipine hydrochloride.
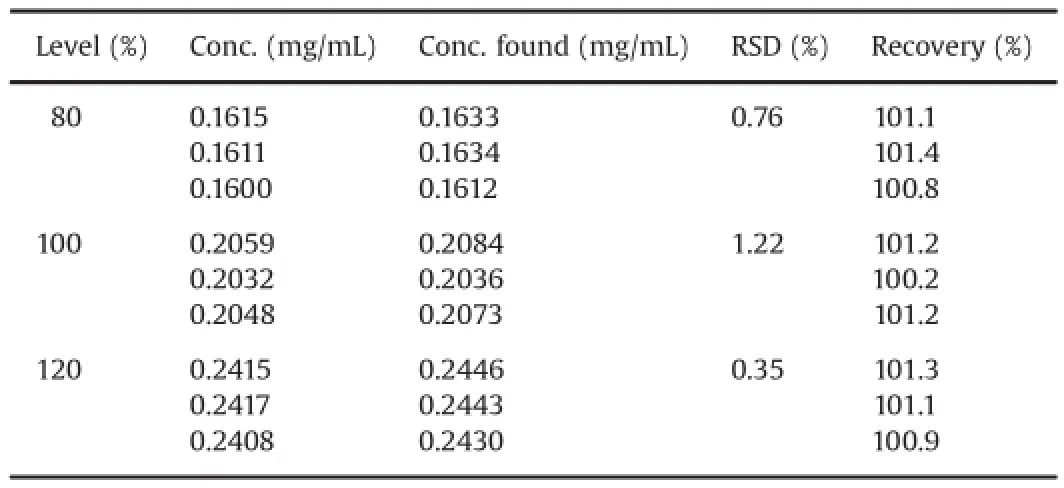
Table 5Accuracy studies for benidipine hydrochloride UPLC assay method.
4.Conclusion
In conclusion,all process related impurities of benidipine hydrochloride produced according to the synthetic route given in Scheme 1 were identified,synthesized and characterized.

Fig.5.Assay chromatograms(UPLC)of stress-testing studies:(A)untreated sample,(B)thermal,(C)day-light,(D)UV-light,(E)neutral hydrolysis(boiling),(F)acidic hydrolysis(HCl),(G)basic hydrolysis(NaOH),and(H)oxidation(H2O2).

Table 6Stress-testing and stability studies of benidipine.
Structural elucidations of these compounds were done using MS,IR and NMR(1H,13C,DEPT)spectral data.Thus,the regulatory requirement has been fulfilled by characterizing all those impurities and the prepared impurity standards have beenusedduringanalyticalmethoddevelopmentandvalidation studies.This work also supported the optimization stage of the process development and enabled one to see the critical points of the process.Using the knowledge of the impurity formation pathways,benidipine production process was carefully optimized to eliminate or minimize the formation of impurities,and the amounts of all impurities described in this article were successfully reduced below 0.10%in the final drug substance.A separate HPLC method was developed and validated for determination of Ben-2 in benidipine drug substance. Due to the weakness of the known potentiometric titration assay method for analysis of the stability samples,a new stability-indicating UPLC method was developed,validated and used during analyses of stability samples of benidipine.Stability studies were conducted under stress,accelerated and longterm stability conditions,and benidipine was found sensitive to oxidizing agents and stable under normal conditions.
Acknowledgments
The authors are thankful to the management of Deva Holding A.S.,Istanbul,Turkey,for supporting this work and the Scientific and Technological Research Council of Turkey(TUBITAK-TEYDEB Project no:3110426)for the financial support.
Appendix A.Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.jpha.2015.02.001.
References
[1]K.Yao,K.Nagashima,H.Miki,Pharmacological,pharmacokinetic,and clinical properties of benidipine hydrochloride,a novel,long-acting calcium channel blocker,J.Pharmacol.Sci.100(2006)243-261.
[2]M.Takayama,M.Matsubara,E.Arakawa,et al.,Comparison of the antiatherosclerotic effects of dihydropyridine calcium channel blocker and HMGCoA reductase inhibitor on hypercholesterolemic rabbits,Vasc.Pharmacol.46(2007)302-308.
[3]H.Suzuki,E.Ono,H.Ueno,et al.,Physico-chemical properties and stabilities of the highly potent calcium antagonist benidipine hydrochloride,Arzneim. Forsch./Drug Res.38(1988)1671-1676.
[4]J.Prous,J.Castaner,KW-3049,Drugs Future 11(1986)936-938.
[5]B.Kalyanaramu,K.Raghubabu,Development of new visible spectrophotometric methods for determination of benidipine hydrochloride in bulk and formulations based on oxidative coupling and diazo coupling reactions,Int.J.Pharm.Biol.Sci.1(2011)57-66.
[6]N.Karadas,S.Sanli,M.Gumustas,et al.,Voltammetric and RP-LC assay for determination of benidipine hydrochloride,J.Pharm.Biomed.Anal.66(2012)116-125.
[7]I.Singhvi,S.C.Chaturvedi,Spectrophotometric methods for estimation of benidipine hydrochloride from tablets,Ind.J.Pharm.Sci.60(1998)113-114.
[8]W.Kang,H.Y.Yun,K.H.Liu,et al.,Determination of benidipine in human plasma using liquid chromatography-tandem mass spectrometry,J.Chromatogr.B:Anal.Technol.Biomed.Life Sci.805(2004)311-314.
[9]A.Ishii,K.Nishida,T.Oka,et al.,Sensitive radioreceptor assay of the calcium antagonist benidipine hydrochloride in plasma and urine,Arzneim.Forsch./ Drug Res.38(1988)1733-1737.
[10]S.Akinaga,H.Kobayashi,S.Kobayashi,et al.,Determination of the calcium antagonist benidipine hydrochloride in plasma by sensitive radioimmunoassay,Arzneim.Forsch./Drug Res.38(1988)1738-1741.
[11]Guidance for industry Q3A(R2),Impurities in new drug substances,in:Proceedings of the International Conference on Harmonisation,2008.
[12]Guidance for industry Q3B(R2),Impurities in new drug products,in:Proceedings of the International Conference on Harmonisation,2006.
[13]G.Goverdhan,A.R.Reddy,K.Srinivas,et al.,Identification,characterization and synthesis of impurities of zafirlukast,J.Pharm.Biomed.Anal.49(2009)895-900.
[14]S.Guntupalli,U.K.Ray,N.Murali,et al.,Identification,isolation and characterization of process related impurities in ezetimibe,J.Pharm.Biomed.Anal. 88(2014)385-390.
[15]M.Dousa,J.Srbeka,S.Radl,et al.,Identification,characterization,synthesis and HPLC quantification of new process-related impurities and degradation products in retigabine,J.Pharm.Biomed.Anal.94(2014)71-76.
[16]A.Abiramasundari,R.P.Joshi,H.B.Jalani,et al.,Stability-indicating assay method for determination of actarit,its process related impurities and degradation products:insight into stability profile and degradation pathways,J. Pharm.Anal.4(2014)374-383.
[17]S.Thomas,S.K.Paul,S.C.Joshi,et al.,Identification,synthesis and characterization of an unknown process related impurity in eslicarbazepine acetate active pharmaceutical ingredient by LC/ESI-IT/MS,1H,13C and1H-1H COSY NMR,J.Pharm.Anal.4(2014)339-344.
[18]B.Saini,G.Bansal,Isolation and characterization of a degradation product in leflunomide and a validated selective stability-indicating HPLC-UV method for their quantification,J.Pharm.Anal.(2015),http://dx.doi.org/10.1016/j. jpha.2014.10.003,in press.
[19]P.Sudhakar,M.Nirmala,J.M.Babu,et al.,Identification and characterization of potential impurities of amlodipine maleate,J.Pharm.Biomed.Anal.40(2006)605-613.
[20]R.Rapolu,C.K.Raju,K.Srinivas,et al.,Isolation and characterization of a novel acid degradation impurity of amlodipine besylate using Q-TOF,NMR,IR and single crystal X-ray,J.Pharm.Biomed.Anal.99(2014)59-66.
[21]R.N.Tiwari,N.Shah,V.Bhalani,et al.,LC,MSnand LC-MS/MS studies for the characterization of degradation products of amlodipine,J.Pharm.Anal.5(2015)33-42.
[22]Guidance for Industry Q1A(R2),Stability testing of new drug substances and products,in:Proceedings of the International Conference on Harmonisation,2003.
[23]Stability testing:photostability testing of new drug substances and products Q1B,in:Proceedings of the International Conference on Harmonisation,1996.
[24]Benidipine hydrochloride/official monographs,Japanese Pharmacopoeia,sixteenth edition,.Pharmaceuticals and Medical Devices Agency,Tokyo,Japan,2011,pp.424-425.
[25]A.Dondoni,A.Massi,E.Minghini,et al.,Model studies toward the synthesis of dihydropyrimidinyl and pyridyl alfa-amino acids via three-component Biginelli and Hantzsch cyclocondensations,J.Org.Chem.68(2003)6172-6183.
[26]F.P.Guengerich,W.R.Brian,M.Iwasaki,et al.,Oxidation of dihydropyridine calcium channel blockers and analogs by human liver cytochrome P-450 IIIA4,J.Med.Chem.34(1991)1838-1844.
[27]ICH Q2(R1),Validation of analytical procedures,in:Proceedings of the International Conference on Harmonisation,2005.
11 November 2014
in revised form
http://dx.doi.org/10.1016/j.jpha.2015.02.001
2095-1779/©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
☆Peer review under responsibility of Xi'an Jiaotong University.
.Tel.:+90 282 7581771x4413;fax:+90 282 7581770.
E-mail addresses:ebellur@deva.com.tr,esenbellur@yahoo.com(E.Bellur Atici).
 Journal of Pharmaceutical Analysis2015年4期
Journal of Pharmaceutical Analysis2015年4期
- Journal of Pharmaceutical Analysis的其它文章
- JPA Prize in 2014
- Application of analytical instruments in pharmaceutical analysis
- Simultaneous quantitation of folic acid and 5-methyltetrahydrofolic acid in human plasma by HPLC-MS/MS and its application to a pharmacokinetic study☆
- Multi-spectroscopic investigation of the binding interaction of fosfomycin with bovine serum albumin☆
- Simultaneous determination of four Sudan dyes in rat blood by UFLC-MS/MS and its application to a pharmacokinetic study in rats☆
- Fabrication of multiwalled carbon nanotube-surfactant modified sensor for the direct determination of toxic drug 4-aminoantipyrine☆