
固相萃取-高效液相色谱法测定畜禽肉中复合硝基酚钠残留
2015-11-08 09:21林燕翠唐中令王洪军
食品工业科技 2015年16期
林燕翠,曹 涛,*,唐中令,卢 芳,王洪军
(1.深圳市通量检测科技有限公司,广东深圳618000;2.嘉吉动物蛋白(安徽)有限公司,安徽滁州239000)
固相萃取-高效液相色谱法测定畜禽肉中复合硝基酚钠残留
林燕翠1,曹涛1,*,唐中令1,卢芳1,王洪军2
(1.深圳市通量检测科技有限公司,广东深圳618000;2.嘉吉动物蛋白(安徽)有限公司,安徽滁州239000)
通过碱性溶液提取,再用正己烷萃取除去脂肪,用乙酸调pH为酸性条件后用乙酸乙酯-异丙醇反萃取,浓缩后再通过混合阴离子固相萃取柱净化,最后采用高效液相色谱分离检测。结果:三种复合硝基酚钠组分在0.05~1.0 μg/mL范围内其浓度与峰面积呈线性,相关系数均可达到0.999;通过萃取、反萃取以及固相萃取等多种手段净化,样品基质几乎无干扰,检出限可达到0.003~0.005 mg/kg;通过对0.25、0.5、1.0 μg进行加标测试,其回收率在81%~95%之间,相对标准偏差均小于10%。
固相萃取,高效液相色谱法,复合硝基酚钠
复合硝基酚钠也称为复硝酚钠,其主要成分包括5-硝基愈创木酚钠(5NG)、邻硝基苯酚钠(ONP)和对硝基苯酚钠(PNP),作为一种广谱型植物生长调节剂能够加速动植物生长[1-3],提高产量,既可单独使用,也可以作为增效剂与农药、肥料、饲料等配合使用,在农药以及养殖业领域适用非常广泛。然而,由于该类物质含有高致毒的苯酚基团和硝基苯基团,能够促使细胞变性、蛋白凝固,对皮肤具有强烈刺激作用,且会在环境中富集,对人以及环境的危害非常大[4-6]。我国在2002年已经通过农业部第193号公告明确规定禁止在所有用途和所有食用动物中添加硝基酚钠,同年在农业部第235号公告中要求所有食用动物中不得检出该类物质。目前对复合硝基酚钠的检测标准仅有针对出口蔬菜采用GC-ECD方法检测[7]以及对复合硝基酚钠原药组分以及饲料的检测文献[8-11],而对于不得添加和不得检出的动物性食品并没有国家或行业的标准方法,并且相关文献也参考GC-ECD方法对鱼虾类产品进行分析[12],然而该方法由于前处理复杂,衍生化对试剂和条件要求苛刻,方法灵敏度和精密度存在一定不足。本文以最具有代表性的动物性食品-畜禽肉为基质,根据复硝基酚钠组分在不同pH溶解性差异用碱溶液提取,正己烷除去脂肪,酸性条件下用乙酸乙酯-异丙醇混合溶剂反萃取。再结合其结构特性,利用硝基苯酚结构的强吸电子基团与混合阴离子固相萃取柱填料的季铵盐结构选择性结合的特点,选择用混合阴离子固相萃取柱净化,再用高效液相色谱-紫外检测器完成畜禽肉类食品的定量检测[13],以期为动物性食品中复合硝基酚钠残留的检测提供检验依据,从而规范复合硝基酚钠的使用,为动物性食品安全添砖献瓦。
1 材料与方法
1.1材料与仪器
甲醇、正己烷、乙酸乙酯、甲酸、异丙醇均为色谱纯均购自JT·Baker;盐酸为优级纯购自JT·Baker;氢氧化钠、氯化钠、氨水为分析纯购自上海国药集团化学试剂有限公司;纯水实验室自制一级实验用水。
LC-20A液相色谱仪,配备紫外检测器日本岛津公司;HP5016SY型氮吹仪上海济成分析仪器有限公司;LJX-ⅡB型离心机上海安亭科学仪器;pH计意大利哈纳;Cleanert PAX混合型阳离子固相萃取柱500 mg/6 mL,购自天津博纳艾杰尔科技有限公司,使用前依次用3 mL水和3 mL 5%氨水甲醇溶液活化;对硝基苯酚、邻硝基苯酚、2-甲氧基-5硝基苯酚均为分析标准品均购自阿拉丁试剂公司,纯度均大于99.5%。
1.2实验方法
1.2.1复合硝基酚钠的提取畜禽肉类样品去骨后经过组织捣碎机制备成肉糜,称取肉糜样品2.0 g于50 mL离心管中,加入0.5 mol/L氢氧化钠水溶液10 mL,振荡提取10 min后4000 r/min离心3 min,取上清液于另一套50 mL离心管中,加入10 mL正己烷,振荡后以4000 r/min离心3 min,上层溶液弃去。用3 mol/L的盐酸调整pH至3.0,再加入5.0 g氯化钠,振荡,加入15 mL乙酸乙酯-异丙醇(4∶6)混合溶剂混匀,振荡后4000 r/min离心5 min,取上层溶液于15 mL离心管中,50℃下氮吹至约1 mL,待净化。
1.2.2提取溶液的净化向样品提取液中加入3 mL含5%氨水的甲醇溶液,通过已活化好的混合阳离子固相萃取柱,再加入3 mL水溶液淋洗,抽干,用5 mL 2%甲酸甲醇溶液洗脱,洗脱液50℃氮吹至约0.5 mL(不得吹干),用流动相定容至2 mL,0.45 μm滤膜过滤待测。
1.2.3检出限的测定对浓度各为0.01 μg/mL的三种混合标液重复进样7次,根据其峰面积计算7次测定结果的平均峰面积以及标准偏差,根据检出限计算公式,得到仪器检出限DL。本次实验过程称量样品为2.0 g,定容体积为2.0 mL,根据仪器检出限换算成方法检出限MDL。
1.3测定条件
色谱柱:Venusil ASB C18色谱柱,4.6 mm×250 mm,5 μm;流动相为甲醇∶pH=3.0水(55∶45,v/v);柱温:30℃;检测波长:0~10 min波长为320 nm,10 min后波长为275 nm;进样量:20 μL。
1.4结果计算
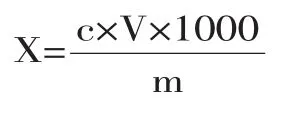
式中,X—试样中5NP,PNP,ONP的含量,mg/kg;c—样品溶液代入标准曲线得到的浓度值含量,μg/mL;V—样品最终定容体积,mL;m—样品称样量,g。
2 结果与讨论
2.1色谱图及流动相的选择
由于待测组分在偏酸性条件下才不会被电离,从而能被C18色谱柱有效保留,所以考虑水相流动相应该调整为弱酸性,实验固定其他条件,调整pH从2.0~6.0进行对三种物质进行测定,检测发现pH在2.0时出峰提前一点,5NP和PNP的峰型略有重叠,分离度略差。而当pH大于5时峰宽增加,推测为pH增加导致溶液H+浓度下降,使组分电离平衡向电离方向便宜,在色谱柱中分配不集中,导致峰宽增加。而pH在3.0~4.0之间时出峰时间和峰型变化不大,但pH=3.0时灵敏度略高一点,因此本实验用磷酸调pH=3.0,有机相采用甲醇。
当水相比例35%时,出峰较快,峰型好,但是ONP和PNP的分离度只有1.1,不能有效分离;当水相比例在55%时,5NP和PNP的分离度虽然有所提高,但是ONP的出峰时间延迟到20min后,峰型较差,容易出现拖尾,因此本实验采用45%的水相作为检测流动相,分离度都大于1.5,而且峰型尖锐,比较适合定量分析。结果如图1所示,其中5NP保留时间为6.99 min;PNP保留时间为7.48 min;ONP保留时间为12.55 min。
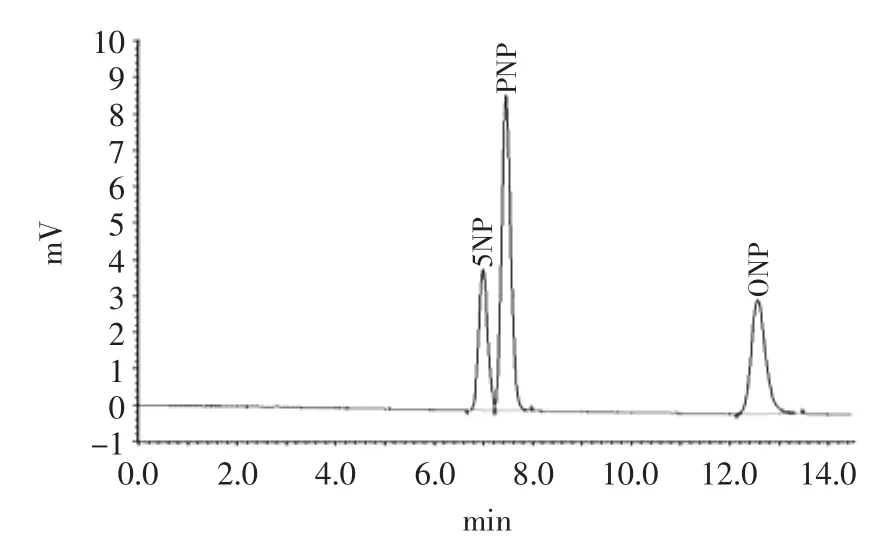
图15 NP、PNP和ONP标准溶液的色谱图Fig.1 The standard solution chromatogram of 5NP,PNP and ONP
2.2检测波长选择
采用二极管阵列检测器对ONP、PNP以及5NP进行了波长扫描,如图2所示,5NP最大吸收波长为343 nm,PNP最大吸收波长为317 nm,ONP最大吸收波长为275 nm,由于5NP和PNP出峰时间靠近,无法变波长进行检测,综合考虑选择320 nm作为检测波长,这样二者均有较大吸收,而ONP可以选择最大吸收波长275 nm作为检测波长。因此本实验0~10 min采用320 nm,10 min之后275 nm作为检测波长。
2.3前处理分析
由于复合硝基酚钠的三种组分在碱性条件下均以负离子形式存在,容易溶解在水相中,因此本实验采取碱溶液提取,不仅能够阻止大部分水不溶性的杂质,而且强碱性能够使蛋白质变性形成沉淀,通过离心除去,而微溶于水中的脂肪颗粒则可以通过正己烷萃取除掉。通过调整酸性条件后,三种组分呈现分子形态,能在有机溶剂中富集;加入氯化钠饱和水相能进一步降低待测物在水相中残留,提高提取效率。由于三种组分均具有一定挥发性,不适合旋转蒸发;另外,通过标准品验证,氮吹干后再复溶,目标物损失50%以上,尤其是ONP几乎损失100%,因此在氮吹时也不能吹干,至少应保留0.5 mL溶剂(如图3、图4所示)。
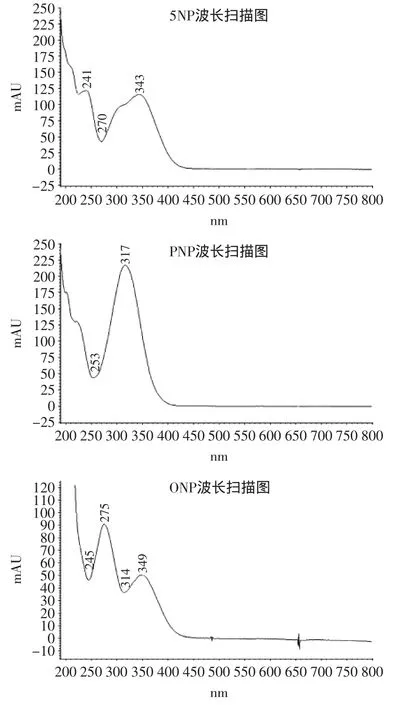
图25 NP、PNP和ONP波长扫描图Fig.2 The wavelength scan of 5NP,PNP and ONP

图3 1.0 μg/mL标准溶液氮吹至约0.5 mL复溶Fig.3 1.0 μg/mL standard solution nitrogen to about 0.5 mL dissolution

图4 1.0 μg/mL标准溶液氮吹干复溶Fig.4 1.0 μg/mL standard solution of nitrogen to dry dissolution
2.4线性范围及相关系数
对浓度范围0.05~1.0 μg/mL混合标准溶液进行二元线性拟合,根据表1所示,PNP、ONP和5NP三种组分含量以及峰面积呈线性关系,其中5NP线性回归方程Y=45231X+30,R2=0.9999;PNP线性回归方程Y=90346X-150,R2=0.9999;ONP线性回归方程Y= 60040X-523,R2=0.9998。
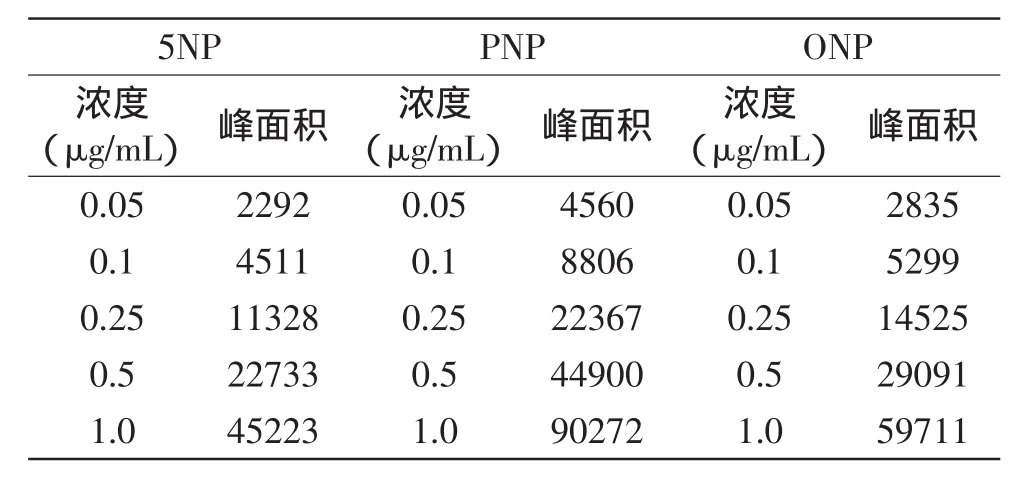
表1 标准曲线记录表Table 1 The record of standard curve
2.5方法检出限
本实验通过对较低浓度(各0.01 μg/mL)的三种混合标液重复进样7次,其峰面积如表2所示,5NP、PNP以及ONP三种组分检出限分别为0.005、0.003和0.004 mg/kg。

表2 检出限峰面积表Table 2 The detection limit of peak area
2.6方法回收率及相对标准偏差
对检测为阴性的猪肉和鸡肉绞碎后分别称取2.0 g,分别加入0.5、1、2 μg,相同浓度加标重复进行3次实验,其加标回收率以及相对标准偏差如表3所示,回收率都在81%~95%范围内,相对标准偏差都小于10%。

表3 加标回收率以及相对标准偏差实验结果表Table 3 The result of recoveries and relative standard deviation
2.7样品分析
按照本方法对市售猪肉和鸡肉分别抽检10份,结果见图5、图6,其中一份猪肉检出ONP,检出结果为0.11 mg/kg,其余均为未检出。

图5 鸡肉样品色谱图Fig.5 The Chromatogram of Chicken
3 结论
本方法通过实验研究,采用阴离子固相萃取柱净化,高效液相色谱法测定畜禽肉中复合硝基酚钠的残留,提取简单,回收率均在81%~95%范围内;该方法净化方式针对性强,杂质去除彻底,从而能够提高灵敏度,5NP、PNP以及ONP三种组分检出限分别为0.005、0.003、0.004 mg/kg;本方法定量结果准确,在0.05~1.0 μg/mL浓度范围内线性相关系数均大于0.999;实验重复性好,相对标准偏差均小于10%,本文为畜禽肉中复合硝基酚钠的残留检测提供了一定依据。

图6 猪肉样品色谱图Fig.6 The Chromatogram of Chicken
[1]王美桂.新型植物生长调节剂——复硝酚钠[J].中国农业科技,2006(7):19.
[2]赵康,陈其武,杨延杰.不同浓度复硝酚钠处理对番茄穴盘苗的影响[J].北方园艺,2012(3):5-7.
[3]于彩莲,刘波,燕红,等.复硝酚钠及其组分对大豆种子萌发的影响[J].大豆科学,2010(3):440-443.
[4]孙炳剑,郑先福,郑昊.复硝酚钠急性毒性的初步研究[J].河南农业大学学报,2007(1):73-76.
[5]郑先福,孙炳剑,郑昊.复硝酚钠的致突变性研究[J].河南农业大学学报,2007(4):94-96.
[6]赵光辉.谈挥发性酚对渔业的危害 [J].内陆水产,1994(Z1):35-36.
[7]中国人民共和国国家进出口商品检验局.SN 0193-93出口蔬菜中复硝盐残留量检测方法[S].北京:标准出版社,1993.
[8]杨晶,曹立冬,王胜翔,等.复硝酚钠原药中3种有效成分的高效液相色谱分析[J].农药学学报,2013(4):439-444.
[9]卫晓红,宋吉利,殷刚,等.高效液相色谱法测定复硝酚钠的三种组分[J].理化检验(化学分册),2011(3):331-332.
[10]刘萍萍,黄修柱,刘绍仁.毛细管气相色谱法分析复硝酚钠水剂[J].农药科学与管理,1998(S1):10-12.
[11]邢丽红,孙伟红,翟毓秀,等.高效液相色谱法检测饲料中的复硝酚钠[J].中国饲料,2013(2):42-44.
[12]邢丽红,冷凯良,孙伟红,等.气相色谱法检测鱼和虾中复硝酚钠残留量[J].中山大学学报,2013(6):104-109.
[13]王芳,岳方.固相萃取-高效液相色谱法同时测定水中硝基苯酚的三种异构体[J].广东化工,2011(10):155-156.
Solid phase extraction-determination of nitrophenols sodium residue in meat of livestock and poultry by HPLC
LIN Yan-cui1,CAO Tao1,*,TANG Zhong-ling1,LU Fang1,WANG Hong-jun2
(1.Shenzhen Total Test Technology Co.,Ltd.,Shenzhen 618000,China;2.Cargill Animal Protein(Anhui)Co.,Ltd.,Chuzhou 239000,China)
This method gave an account that extracted by alkaline solution,extracted by hexane to remove the fat,back-extraction with ethyl acetate-isopropanol under acid condition,purified by mixing anionic solid-phase extraction(SPE),finally,using liquid chromatography(HPLC)for testing.The three kinds of compound sodium nitrophenol components concentration and the peak area was linear correlation within the scope of 0.05~1.0 μg/mL.The correlation was 0.999.Through the extraction,back extraction and a variety of means such as solid phase extraction purification,the sample almost had no interference,so the detection limit could reached 0.003~0.005 mg/kg.Through the standard addition test of 0.25,0.5,1.0 μg,its recovery was between 81%~95%,relative standard deviation was less than 10%.
mixing anionic solid-phase extraction(SPE);HPLC;compound nitrophenols sodium
TS251.7
A
1002-0306(2015)16-0082-04
10.13386/j.issn1002-0306.2015.16.008
2014-10-31
林燕翠(1988-),女,本科,主要从事畜禽及水产等动物性食品兽药残留检测方面的研究,E-mail:cuill1234@sina.com。
曹涛(1987-),男,本科,主要从事畜禽及水产等动物性食品兽药残留检测方面的研究,E-mail:vitccy@163.com。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
当代水产(2022年4期)2022-06-05
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
化工管理(2021年7期)2021-05-13
中国农资(2016年1期)2016-12-01
化工进展(2015年3期)2015-11-11
河北工业科技(2015年4期)2015-02-27
现代检验医学杂志(2015年1期)2015-02-06
