
甲型流感病毒感染过程中干扰素介导的天然免疫应答机制
2015-06-05 09:35陈超池晓娟白庆玲陈吉龙
生物工程学报 2015年12期
陈超,池晓娟,白庆玲,陈吉龙,2
甲型流感病毒感染过程中干扰素介导的天然免疫应答机制
陈超1,池晓娟1,白庆玲1,陈吉龙1,2
1 福建农林大学动物科学学院,福建福州 350002 2 中国科学院微生物研究所中国科学院病原微生物与免疫学重点实验室,北京 100101

陈超, 池晓娟, 白庆玲, 等. 甲型流感病毒感染过程中干扰素介导的天然免疫应答机制. 生物工程学报, 2015, 31(12): 1671–1681.Chen C, Chi XJ, Bai QL, et al. Mechanisms underlying interferon-mediated host innate immunity during influenza A virus infection. Chin J Biotech, 2015, 31(12): 1671–1681.
甲型流感病毒作为引起人类和动物急性呼吸道传染病的一个主要病原体,在世界范围内广泛流行。研究表明,甲型流感病毒感染宿主后会诱导宿主的天然免疫应答。甲型流感病毒感染可引起Toll样受体(Toll like receptors,TLRs) 和RIG-I样受体(RIG-I like receptors,RLRs) 等宿主模式识别受体介导的抗病毒信号通路的活化,并在多种机制调控下诱导干扰素和其他细胞因子的表达,如Ⅰ型干扰素、Ⅲ型干扰素等,从而启动干扰素刺激基因 (Interferon stimulated genes,ISGs) 的转录及其抗病毒蛋白的表达,进而实现抗病毒作用。本文就甲型流感病毒感染与干扰素介导的天然免疫应答相关的信号通路和调控机制进行综述。
甲型流感病毒,天然免疫,模式识别受体,干扰素,干扰素刺激基因
流感病毒属于正黏病毒科 (Orthomyxoviridae)的单股负链RNA病毒[1-2]。按照病毒的核蛋白 (Nuclear protein,NP) 和基质蛋白 (Matrix protein,MP) 抗原性的不同可分为A (甲)、B (乙)、C (丙) 三型。根据病毒囊膜表面糖蛋白血凝素 (Hemagglutinin,HA) 和神经氨酸酶 (Neuraminidase,NA) 的差异,流感病毒可分为许多亚型。流感病毒基因组为分节段形式存在,使病毒的基因组容易发生重排,并在药物和疫苗等免疫压力下容易发生突变[3]。流感病毒以甲型流感病毒 (Influenza A virus,IAV) 带来的危害最大,最典型的例子是发生于1918年的“西班牙大流感”,造成了近5 000万人的死亡[4]。因此,流感是一种严重危害人类健康的疫病,而且也造成了重大的经济损失[3]。
甲型流感病毒感染宿主后,宿主天然免疫系统作为抵抗病毒入侵的第一道防线,会立即被激活并启动相应的免疫应答机制。天然免疫应答的激活首先主要通过宿主3种模式识别受体 (Pattern recognition receptors,PRRs) 对病原相关模式分子 (Pathogen-associated molecular patterns,PAMP) 进行特异性识别。研究发现,流感病毒的基因组为含5ʹ三磷酸的RNA (5ʹ-triphosphate RNA),在其复制过程中会产生双链RNA,这些核酸物质是流感病毒诱导宿主天然免疫应答的主要病原相关模式分子,而识别该类PAMP的宿主模式识别受体主要有Toll样受体和RIG-I样受体等[5-6]。流感病毒入侵的信号被识别后,宿主细胞内PRRs介导的天然免疫信号通路被激活,这些通路调控宿主干扰素 (Interferon,IFN) 等细胞因子基因的转录,从而分泌相关细胞因子如Ⅰ型干扰素和Ⅲ型干扰素。
在天然免疫应答中产生的干扰素与相应的干扰素受体结合,激活Janus酪氨酸激酶-信号转导子和转录活化子 (Janus tyrosine kinases- signal transducers and activators of transcription,JAK-STAT) 信号通路。活化的JAK-STAT信号通路启动一系列干扰素刺激基因的表达,而这些干扰素刺激基因的产物是非常重要的一类抗病毒分子,不仅调节着机体的免疫功能,而且在抗病毒感染过程中扮演着重要角色[7-9](图1)。甲型流感病毒感染所引起天然免疫应答的识别、调控和信号传导等过程错综复杂,这方面的研究方兴未艾。
1 参与抗甲型流感病毒天然免疫的重要模式识别受体
1.1 TLRs介导的抗甲型流感病毒天然免疫应答
TLRs作为识别PAMP的一类重要受体,早在1996年就被报道在免疫系统中扮演着重要角色[10]。TLRs在多种细胞中都有表达,目前在哺乳动物发现的TLRs有13种 (TLR1-13),识别病原微生物脂多糖、核酸等成分[11]。TLRs表达于细胞表面和细胞内部。其中TLR3、TLR7/8、TLR9等主要是在细胞内的一些膜结构上对病毒核酸的识别,而其他TLRs则主要识别一些非核酸的PAMP[12]。流感病毒作为单链RNA病毒,其复制过程中产生的中间产物包含双链RNA,所以当其感染宿主后,流感病毒的双链和单链RNA都存在于细胞中。在宿主细胞内识别流感病毒核酸的TLRs主要为TLR3和TLR7/8[13-14]。
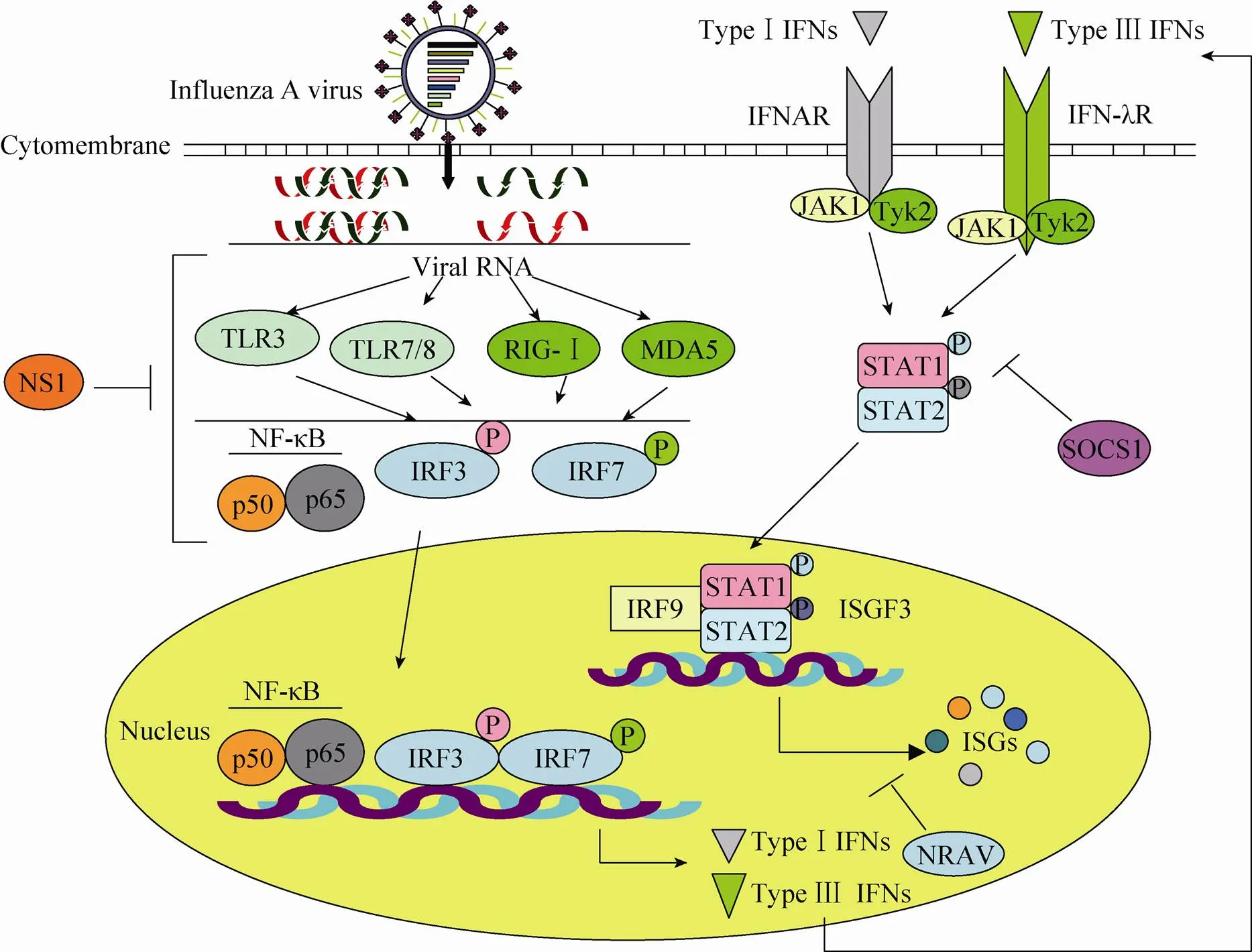
图1 甲型流感病毒感染诱导的宿主细胞天然免疫应答通路
Kawai等研究表明,RNA病毒感染宿主后,病毒核酸会被TLR3所识别,活化转录因子IRF3 (IFN regulatory factor 3,IRF3)、AP1 (Activator protein 1,AP1) 和NF-κB的p50/p65,启动干扰素的表达,进而激活干扰素介导的天然免疫应答,抵抗病毒的感染[15]。最近有研究表明,TLR3和RIG-I都被敲低 (Knockdown) 后,甲型流感病毒感染人肺泡上皮细胞所引起的干扰素诱导表达会被抑制[16]。有趣的是,Le Goffic等研究发现,TLR3敲除小鼠在感染甲型流感病毒后,小鼠体内炎症介质的水平相比于野生型小鼠低,因而获得了一定的生存优势[13],这些结果表明TLR3参与了抗流感病毒天然免疫应答的调节过程。其他研究也表明了TLR3在调节宿主炎症反应过程中发挥重要作用[17]。
TLR7/8主要识别病毒的ssRNA[18]。当流感病毒感染宿主后,TLR7/8介导中性粒细胞的激活[19],并且宿主体内会表达大量的Ⅰ型干扰素和Ⅲ型干扰素。TLR7识别流感病毒的ssRNA后,激活MyD88依赖的信号通路,活化IRF7,从而诱导产生Ⅰ型干扰素和炎性因子,同时也激活转录因子NF-κB信号通路,诱导干扰素的表达,来抵抗流感病毒的感染和复制[14,20-21]。有趣的是,Panq等研究发现,宿主在感染低滴度的甲型流感病毒后,TLR7与RIG-I介导的炎症过程促进了病毒的复制[22]。其研究发现流感病毒感染细胞后,通过TLR7与RIG-I信号通路介导的炎症反应,招募了大量易被甲型流感病毒感染的靶细胞到呼吸道,从而提高了感染部位的病毒载量。流感病毒感染与宿主天然免疫应答的互作关系错综复杂,TLRs作为天然免疫应答的重要模式识别受体发挥多方面的作用,而其中的内在关系与具体作用机制还有待于进一步阐明。
1.2 RLRs介导的抗甲型流感病毒天然免疫应答
RLRs家族主要成员包括视黄酸诱导基因Ⅰ(Retinoic acid-inducible geneⅠ,RIG-Ⅰ) 的表达蛋白、黑色素瘤分化相关蛋白5 (Melanoma differentiation-associated protein 5,MDA5)、遗传学和生理学实验室蛋白2 (Laboratory of genetics and physiology 2,LGP2)[23-24]。RLRs家族的结构有相似之处,都含有DExD/H解旋酶结构域以及C末端结构域。RIG-Ⅰ和MDA5的N端含有2个CARD (Caspase recruitment and activation domain,CARD) 结构域,并且RIG-Ⅰ的C末端结构域中含有使RIG-Ⅰ在无刺激物时处于一种抑制状态的抑制性结构域 (RD结构域)[25-28]。RIG-Ⅰ对于5ʹ端含有3个磷酸基团结构的RNA的识别更敏感,MDA5则倾向于识别长双链RNA[29-30]。流感病毒感染宿主细胞后,对其病毒RNA进行识别的RLRs主要有RIG-I和MDA5[24]。
研究表明,RIG-Ⅰ和MDA5在宿主天然免疫中的作用至关重要。RIG-Ⅰ或MDA5缺陷的小鼠都表现出对RNA病毒的易感性[31]。流感病毒感染宿主细胞后,其RNA会被RIG-Ⅰ和MDA5分别识别[32-33],激活天然免疫应答的信号通路。这些RLRs感应流感病毒感染后,其CARD结构域招募重要的接头蛋白MAVS (VISA/IPS-I/Cardif),随后激活转录因子IRF3、IRF7和NF-κB,进而诱导干扰素和细胞因子的表达,抵抗病毒的 感染[34-35]。
有研究表明,MDA5需在LGP2的帮助下才可继续下游信号的传递[36]。此外,Wei等研究发现,MDA5 CARD结构域的招募和Caspase的激活依赖IRF7信号通路[37],过表达的CARD可强烈激活IFN-β启动子、上调抗病毒分子 (如OAS、PKR和MxA) 和炎症因子 (如IL-2、IL-6、IFN-α和IFN-γ) 的表达。
2 干扰素介导的抗甲型流感病毒天然免疫
1957年科学家Isaacs和Lindenmann利用鸡胚绒毛尿囊膜研究流感病毒干扰现象时发现一种细胞因子,由于其具有干扰病毒复制的功能,而被命名为干扰素[38],后被证实为Ⅰ型干扰素。之后又陆续发现了Ⅱ型干扰素和Ⅲ型干扰素。IFN最重要的功能是参与宿主抗病毒感染过程[39],其抗病毒作用具有广谱性,其中Ⅰ型干扰素和Ⅲ型干扰素在抗甲型流感病毒天然免疫应答中起到重要的作用。
2.1 Ⅰ型干扰素介导的抗甲型流感病毒天然免疫应答
如上所述,宿主模式识别受体感应流感病毒的核酸,激活了由PRR介导的信号通路,诱导产生大量的Ⅰ型干扰素,它们是宿主抵抗流感病毒感染机制中非常重要的一类免疫因子[40-41]。Ⅰ型干扰素种类较多,其中IFN-α和IFN-β是非常关键的Ⅰ型干扰素[42]。当流感病毒感染宿主细胞后诱导产生Ⅰ型干扰素,干扰素通过自分泌、旁分泌等形式与靶细胞上的干扰素受体 (IFNAR1和IFNAR2) 结合[7],使受体发生二聚化,从而导致受体空间构象发生变化。这样,与受体C端结合的JAK (主要是JAK1和Tyk2) 激酶由于相互作用而发生磷酸化。活化的JAK激酶使下游的STAT1和STAT2磷酸化[8,43],进而形成二聚体,二聚化的STAT蛋白空间构象发生变化导致STAT核定位信号暴露,其介导STAT进入细胞核。细胞核内的STAT作为转录因子与IRF9形成一个三分子复合物,即形成了干扰素刺激基因因子3 (IFN-stimulated gene factor 3,ISGF3)。ISGF3与相关基因上的干扰素刺激反应元件 (IFN-stimulated response elements,ISREs) 结合后,启动这些基因的大量转录。这些基因包括数以百计的干扰素刺激基因,控制了大量抗病毒分子的表达,从而抵抗病毒的感染[44-45]。关于Ⅰ型干扰素在抗甲型流感病毒天然免疫中的作用已积累了大量的研究数据,近年来,针对Ⅰ型干扰素抗甲型流感病毒的信号通路是研究的热点。
2.2 Ⅲ型干扰素介导的抗甲型流感病毒天然免疫应答
相比于Ⅰ型干扰素,Ⅲ型干扰素发现较晚,是在2003年由两个独立的实验室基于基因组序列鉴定的一种新型干扰素家族[46-47]。流感病毒感染宿主细胞后,RIG-I、MDA5、TLR3、TLR7/8等受体介导的信号通路会引起大量Ⅲ型干扰素的分泌[48-49],其中RIG-I起重要的作用。Ⅲ型干扰素是宿主抵御流感病毒感染第一道屏障的重要成员[48,50]。
人类Ⅲ型干扰素家族包括3个成员,即IL-29 (也称IFN-λ1)、IL-28A (也称IFN-λ2) 和IL-28B (也称IFN-λ3)[46]。宿主模式识别受体感应流感病毒感染后所激活的信号通路及其调控Ⅲ型干扰素的表达过程与Ⅰ型干扰素的表达调控机制很相似。病毒感染诱导产生的Ⅲ型干扰素与其受体结合后,启动的是与Ⅰ型干扰素相似的信号通路,即通过激活JAK-STAT信号转导通路[8,51],磷酸化的STAT蛋白进入细胞核内形成ISGF3复合物,随后ISGF3与ISREs结合,从而诱导多种ISGs的大量表达[45,52],抑制流感病毒感染。Holzinger等研究证实,宿主在感染甲型流感病毒后,诱导高水平的Ⅲ型干扰素表达,后者刺激重要的抗病毒蛋白MxA的产生,进而MxA阻断流感病毒的复制[53]。此外,研究表明,IFN-λ1预处理的肺泡Ⅱ型上皮细胞在甲型流感病毒感染后,相关的ISGs (主要为OAS、MxA和ISG56) 表达会上调,这些ISGs通过作用于流感病毒蛋白等方式,干扰流感病毒的复制过程[54]。
如上所述,干扰素所激活的JAK-STAT通路在抗病毒免疫应答中发挥至关重要的作用,这一通路也受到复杂机制的调控。其中,细胞因子信号抑制蛋白 (Suppressor of cytokine signaling,SOCS) 家族就是一类非常关键的JAK-STAT通路的负调控蛋白。近期,我们研究发现,宿主细胞在感染甲型流感病毒后,通过RIG-I依赖的信号通路,引发肺上皮细胞大量表达Ⅲ型干扰素IFN-λ,而IFN-λ激活JAK-STAT信号通路,诱导天然免疫应答[55]。重要的是,我们研究发现甲型流感病毒能够直接诱导JAK-STAT信号通路的负调控因子SOCS1的大量表达,而SOCS1蛋白通过抑制IFN-λ激活的JAK-STAT通路,从而有利于流感病毒逃脱宿主天然免疫应答 (图1)。有趣的是,通过体外细胞系实验和体内动物模型,我们均发现干扰SOCS1表达或高表达持续活化型的STAT1可显著地降低IFN-λ的表达水平。深入研究发现,甲型流感病毒诱导的SOCS1高表达,可以进一步活化NF-κB。这些结果揭示了甲型流感病毒感染过程中,IFN-λ下游的JAK-STAT信号通路受病毒直接诱导的SOCS1抑制后,宿主代偿性激活NF-κB从而引发了IFN-λ的过量表达,导致恶性的病理反应[55]。这些结果预示着流感病毒感染导致某些细胞因子过量表达的潜在机理。这一机理在一定程度上解释了流感病毒引发高细胞因子血症的分子基础。
2.3 ISGs在抗甲型流感病毒天然免疫中的作用
Ⅰ型干扰素和Ⅲ型干扰素与其相应的受体结合激活了细胞因子信号通路,最终诱导多种ISGs的大量表达,这些ISGs起到了直接抵抗流感病毒感染的作用[45]。近年来的大量研究发现,Ⅰ型干扰素和Ⅲ型干扰素所诱导的ISGs种类繁多。例如,RNA激活的丝氨酸/苏氨酸蛋白激酶 (RNA-activated protein kinase,PKR)、2′-5′-寡腺苷酸合成酶 (2′-5′-oligoadenylate synthetase,OAS) 基因家族、MxA和干扰素诱导跨膜蛋白(Interferon induced transmembrane protein,IFITM) 家族等[46,56-58]。这些ISGs在流感病毒复制的各个环节中发挥着调控作用,并且有的ISGs也参与了流感病毒感染后的宿主细胞凋亡和宿主对PAMPs的识别等过程。
对PKR的研究历史较长,结论也较清晰。现已表明,PKR通过磷酸化翻译起始因子eIF2a,从而阻断病毒蛋白合成[59]。OAS则是激活核糖核酸内切酶 (RNaseL)[60],降解病毒RNA,从而抑制病毒蛋白的合成。MxA蛋白属于GTPase家族,可以特异性地作用于流感病毒NP蛋白,进而抑制病毒的复制[61]。IFITM家族则是近年来的研究热点,2009年Brass等[62]通过siRNA干扰IFITM3基因,发现敲低IFITM3的表达可以明显促进流感病毒的复制,相反,过表达IFITM1、IFITM2或IFITM3都可以抑制流感病毒感染,这些研究证明了IFITM家族抗流感病毒的功能。之后,来自不同实验室的研究也证实IFITM3通过阻碍流感病毒的早期侵入宿主细胞过程,阻断病毒与宿主细胞内涵体膜融合,阻断vRNP的入核及复制,从而起到抵御流感病毒感染的作用[63-65]。
由于ISGs种类繁多,ISGs的表达调控机制也很复杂。有趣的是,最近我们研究发现长链非编码RNA (Long non-coding RNA,lncRNA) 在ISGs表达调控及抗病毒天然免疫反应中具有重要的作用。首先,我们从转录组水平上发现,在感染甲型流感病毒的人肺细胞A549中一种名为NRAV的长链非编码RNA表达水平发生明显降低,进一步研究发现NRAV可通过调控、等多种ISGs基因的组蛋白修饰 (H3K4me3和H3K27me3) 从而抑制这些基因的初始转录 (图1)。这些研究证实了lncRNA NRAV作为ISGs的负调控因子,在流感病毒感染过程中的下调是宿主保护性天然免疫应答机制[66]。这一研究为深入了解流感病毒与宿主的相互作用、阐明流感病毒的致病机理提供重要的科学依据。
3 甲型流感病毒拮抗干扰素介导的天然免疫应答
流感病毒为了拮抗宿主的免疫应答,在长期进化过程中,产生了许多抵抗宿主免疫应答的策略,并可利用或“绑架”宿主的有关成分完成其复制过程[67]。例如,本实验室研究发现,甲型流感病毒感染导致宿主细胞中真核转译起始因子eIF4B水平显著降低,而eIF4B是IFITM3蛋白翻译所必需的,表明了流感病毒通过改变eIF4B水平来下调IFITM3蛋白,以消除其抗病毒作用[65]。此外,我们研究发现,甲型流感病毒可以明显下调宿主细胞的ARHGAP21蛋白水平从而增强了重要的小GTPase Cdc42的活性,而活化的Cdc42协助病毒囊膜蛋白神经氨酸酶向细胞表面转运,促进了病毒复制[67]。
越来越多的证据表明,甲型流感病毒的非结构蛋白NS1在拮抗宿主天然免疫过程中发挥至关重要的作用。例如,流感病毒通过NS1蛋白与TRIM25 (Tripartite motif,一种可激活RIG-I的泛素连接酶) 相结合,抑制了感染细胞中由RIG-I激活的天然免疫信号通路和IFN-β的产生[68-71]。研究表明,NS1可以通过抑制IRF3的激活、干扰IFN的转录、抑制宿主对IFN pre-mRNAs的剪接加工过程等途径来抑制宿主细胞中干扰素的表达,从而达到拮抗干扰素介导的天然免疫[72]。有趣的是,有研究发现宿主感染流感病毒后,病毒NS1蛋白同样会抑制MDA5介导的宿主抗病毒天然免疫信号通路的活化,从而逃脱宿主的天然免疫应答[37]。NS1是一种多功能蛋白,它在拮抗宿主免疫应答中发挥的作用是多方面的 (图1),也是极其复杂的,有关NS1蛋白的作用机制仍有许多问题依然没有准确答案,有待深入研究。
4 小结与展望
综上所述,科学家们对甲型流感病毒感染过程中干扰素介导的天然免疫应答过程及其调控机制有了一定的认识。流感病毒感染宿主细胞后,宿主的PRRs对病毒的PAMP进行识别,激活了以TLRs和RLRs等受体介导的天然免疫应答信号通路,进而诱导高水平的Ⅰ型干扰素、Ⅲ型干扰素和其他细胞因子的分泌,干扰素进一步通过激活JAK-STAT信号通路来调控多种ISGs的表达,最终宿主利用这些抗病毒分子抵御流感病毒的感染与复制。
甲型流感病毒作为一种重要的人畜共患传染病的病原,严重威胁人类健康与危害畜牧业发展。虽然人们对宿主识别流感病毒、宿主天然免疫应答通路及其调控机制有了一定的了解,但是流感病毒-宿主之间的相互作用错综复杂,依然存在着许多问题值得深入探究,如流感病毒感染所诱导的天然免疫与适应性免疫各通路之间的内在关系是什么?以及Ⅰ型干扰素与Ⅲ型干扰素如何协调作用?对流感病毒感染与宿主天然免疫应答的深入研究,将有助于我们全面了解甲型流感病毒的致病机理,并为研发新的抗病毒药物提供有价值的靶点。
[1] von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov, 2007, 6(12): 967–974.
[2] Betakova T. M2 protein-a proton channel of influenza A virus. Curr Pharm Des, 2007, 13(31): 3231–3235.
[3] Lam TT, Wang J, Shen Y, et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature, 2013, 502(7470): 241–244.
[4] Reid AH, Fanning TG, Janczewski TA, et al. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc Natl Acad Sci USA, 2000, 97(12): 6785–6790.
[5] Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature, 2001, 413(6857): 732–738.
[6] Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev, 2009, 22(2): 240–273.
[7] Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol, 2008, 89(1): 1–47.
[8] Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity, 2012, 36(4): 503–514.
[9] von Recum-Knepper J, Sadewasser A, Weinheimer VK, et al. Fluorescence-activated cell sorted-based analysis reveals an asymmetric induction of interferon-stimulated genes in response to seasonal influenza a virus. J Virol, 2015, 89(14): 6982–6993.
[10] Lemaitre B, Nicolas E, Michaut L, et al. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response inadults. Cell, 1996, 86(6): 973–983.
[11] Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol, 2005, 17(1): 1–14.
[12] Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity, 2010, 32(3): 305–315.
[13] Le Goffic R, Balloy V, Lagranderie M, et al. Detrimental contribution of the Toll-like receptor (TLR) 3 to influenza A virus-induced acute pneumonia. PLoS Pathog, 2006, 2(6): e53.
[14] Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science, 2004, 303(5663): 1529–1531.
[15] Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem, 2007, 141(2): 137–145.
[16] Wu WX, Zhang W, Duggan ES, et al. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology, 2015, 482: 181–188.
[17] Le Goffic R, Pothlichet J, Vitour D, et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol, 2007, 178(6): 3368–3372.
[18] Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science, 2004, 303(5663): 1526–1529.
[19] Wang JP, Bowen GN, Padden C, et al. Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood, 2008, 112(5): 2028–2034.
[20] Ludwig S, Ehrhardt C, Neumeier ER, et al. Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J Biol Chem, 2001, 276(14): 10990–10998.
[21] Rahman I, Gilmour PS, Jimenez LA, et al. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem, 2002, 234-235(1/2): 239–248.
[22] Pang IK, Pillai PS, Iwasaki A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Natl Acad Sci USA, 2013, 110(34): 13910–13915.
[23] Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol, 2004, 5(7): 730–737.
[24] Loo YM, Gale Jr M. Immune signaling by RIG-I-like receptors. Immunity, 2011, 34(5): 680–692.
[25] Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol, 2005, 175(5): 2851–2858.
[26] Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol, 2008, 20(1): 17–22.
[27] Komuro A, Horvath CM. RNA-and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol, 2006, 80(24): 12332–12342.
[28] Saito T, Hirai R, Loo YM, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA, 2007, 104(2): 582–587.
[29] Cui S, Eisenächer K, Kirchhofer A, et al. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell, 2008, 29(2): 169–179.
[30] Kato H, Takeuchi O, Mikamo-Satoh E, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med, 2008, 205(7): 1601–1610.
[31] Wang Y, Zhang HX, Sun YP, et al. Rig-I−/−mice develop colitis associated with downregulation of Gαi2. Cell Res, 2007, 17(10): 858–868.
[32] Pichlmair A, Schulz O, Tan CP, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science, 2006, 314(5801): 997–1001.
[33] Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol, 2008, 82(1): 335–345.
[34] Koyama S, Ishii KJ, Kumar H, et al. Differential role of TLR-and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol, 2007, 179(7): 4711–4720.
[35] Schmolke M, García-Sastre A. Evasion of innate and adaptive immune responses by influenza A virus. Cell Microbiol, 2010, 12(7): 873–880.
[36] Satoh T, Kato H, Kumagai Y, et al. LGP2 is a positive regulator of RIG-I-and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA, 2010, 107(4): 1512–1517.
[37] Wei LM, Cui J, Song YF, et al. Duck MDA5 functions in innate immunity against H5N1 highly pathogenic avian influenza virus infections. Vet Res, 2014, 45(1): 66.
[38] Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci, 1957, 147(927): 258–267.
[39] Bazhan SI, Belova OE. Molecular genetic aspects of interferon induction and antiviral action. Vestn Ross Akad Med Nauk, 1998, (3): 18–24.
[40] Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus Res, 2015, 209: 11–22.
[41] Zhu ZX, Yang YF, Wei JC, et al. Type I interferon-mediated immune response against influenza A virus is attenuated in the absence of p53. Biochem Biophys Res Commun, 2014, 454(1): 189–195.
[42] Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol, 2014, 14(1): 36–49.
[43] Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol, 2002, 3(9): 651–662.
[44] Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol, 2006, 8(6): 907–922.
[45] Fensterl V, Sen GC. Interferons and viral infections. Biofactors, 2009, 35(1): 14–20.
[46] Kotenko SV, Gallagher G, Baurin VV, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol, 2003, 4(1): 69–77.
[47] Sheppard P, Kindsvogel W, Xu WF, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol, 2003, 4(1): 63–68.
[48] Jewell NA, Cline T, Mertz SE, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection. J Virol, 2010, 84(21): 11515–11522.
[49] Österlund PI, Pietilä TE, Veckman V, et al. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-λ) genes. J Immunol, 2007, 179(6): 3434–3442.
[50] Lopušná K, Režuchová I, Betáková T, et al. Interferons lambda, new cytokines with antiviral activity. Acta Virol, 2013, 57(2): 171–179.
[51] Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology, 2006, 44(4): 896–906.
[52] Onoguchi K, Yoneyama M, Takemura A, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem, 2007, 282(10): 7576–7581.
[53] Holzinger D, Jorns C, Stertz S, et al. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol, 2007, 81(14): 7776–7785.
[54] Wang JR, Oberley-Deegan R, Wang SL, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-λ1) in response to influenza A infection. J Immunol, 2009, 182(3): 1296–1304.
[55] Wei HT, Wang S, Chen QH, et al. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PLoS Pathog, 2014, 10(1): e1003845.
[56] De Veer MJ, Holko M, Frevel M, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol, 2001, 69(6): 912–920.
[57] Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol, 2008, 8(7): 559–568.
[58] Zhou X, Michal JJ, Zhang LF, et al. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci, 2013, 9(2): 200–208.
[59] Bergmann M, Garcia-Sastre A, Carnero E, et al. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol, 2000, 74(13): 6203–6206.
[60] Min JY, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci USA, 2006, 103(18): 7100–7105.
[61] Dittmann J, Stertz S, Grimm D, et al. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J Virol, 2008, 82(7): 3624–3631.
[62] Brass AL, Huang IC, Benita Y, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell, 2009, 139(7): 1243–1254.
[63] Feeley EM, Sims JS, John SP, et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog, 2011, 7(10): e1002337.
[64] Bailey CC, Zhong GC, Huang IC, et al. IFITM-family proteins: the cell’s first line of antiviral defense. Annu Rev Virol, 2014, 1: 261–283.
[65] Wang S, Chi XJ, Wei HT, et al. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J Virol, 2014, 88(15): 8375–8385.
[66] Ouyang J, Zhu XM, Chen YH, et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe, 2014, 16(5): 616–626.
[67] Wang S, Li H, Chen YH, et al. Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. J Biol Chem, 2012, 287(13): 9804–9816.
[68] Gack MU, Albrecht RA, Urano T, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe, 2009, 5(5): 439–449.
[69] Gack MU, Shin YC, Joo CH, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature, 2007, 446(7138): 916–920.
[70] Mibayashi M, Martínez-Sobrido L, Loo YM, et al. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol, 2007, 81(2): 514–524.
[71] Ayllon J, Garcia-Sastre A. The NS1 protein: a multitasking virulence factor. Curr Top Microbiol Immunol, 2015, 386: 73–107.
[72] Krug RM. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol, 2015, 12: 1–6.
(本文责编郝丽芳)
Mechanisms underlying interferon-mediated host innate immunity during influenza A virus infection
Chao Chen1, Xiaojuan Chi1, Qingling Bai1, and Jilong Chen1,2
1,,350002,,2,,,100101,
Influenza A virus can create acute respiratory infection in humans and animals throughout the world, and it is still one of the major causes of morbidity and mortality in humans worldwide. Numerous studies have shown that influenza A virus infection induces rapidly host innate immune response. Influenza A virus triggers the activation of signaling pathways that are dependent on host pattern recognition receptors (PRRs) including toll like receptors (TLRs) and RIG-I like receptors (RLRs). Using a variety of regulatory mechanisms, these signaling pathways activate downstream transcript factors that control expression of various interferons and cytokines, such as type I and type III interferons. Thus, these interferons stimulate the transcript of relevant interferon-stimulated genes (ISGs) and expression of the antiviral proteins, which are critical components of host innate immunity. In this review, we will highlight the mechanisms by which influenza A virus infection induces the interferon-mediated host innate immunity.
influenza A virus, innate immunity, pattern recognition receptors, interferons, interferon-stimulated genes
June 25, 2015; Accepted:August 3, 2015
Jilong Chen. Tel: +86-10-64807300; Fax: +86-10-64807980; E-mail: chenjl@im.ac.cn
10.13345/j.cjb.150296
Supported by:National Natural Science Foundation of China (No. 31402217).
国家自然科学基金 (No. 31402217) 资助。
2015-08-20
http://www.cnki.net/kcms/detail/11.1998.Q.20150820.0953.002.html
猜你喜欢
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
广东医科大学学报(2020年6期)2020-02-06
现代检验医学杂志(2016年3期)2016-11-15
现代检验医学杂志(2016年1期)2016-11-12
中国继续医学教育(2015年6期)2016-01-07
华南农业大学学报(2015年5期)2015-12-04
哈尔滨医药(2015年6期)2015-12-01
小星星·阅读100分(高年级)(2015年11期)2015-11-28
中国医疗美容(2015年2期)2015-07-19
