
基于不同碱基含量的[FeFe]-氢化酶模型化合物功能化聚合物的合成与表征
2015-06-01 10:30梁绍威詹彩霞李文强王宏丹肖志音刘小明
无机化学学报 2015年1期
梁绍威 钟 伟 詹彩霞 赵 佳 李文强 叶 萍 王宏丹 沈 杰 樊 露 肖志音 刘小明*,,
(1嘉兴学院生物与化学工程学院,嘉兴314001)
(2南昌大学化学系,南昌330031)
基于不同碱基含量的[FeFe]-氢化酶模型化合物功能化聚合物的合成与表征
梁绍威1钟 伟1詹彩霞2赵 佳2李文强2叶 萍1王宏丹1沈 杰1樊 露1肖志音1刘小明*,1,2
(1嘉兴学院生物与化学工程学院,嘉兴314001)
(2南昌大学化学系,南昌330031)
将含二炔基的[2Fe2S]模型化合物[Fe2(μ-SCH2C≡CH)2(CO)6](A)和双叠氮单体2,6-(N3CH2)Py(B,Py=吡啶)与含碱性基团的双炔单体(2-PyCH2)N(CH2C≡CH)2(C,Py=吡啶)通过“点击反应”以不同比例进行共聚,得到了6种含不同碱基量的[FeFe]-氢化酶模型化合物功能聚合物Polymer-1~Polymer-6。通过红外光谱、硫元素分析、热重、扫描电镜和电化学对这些聚合物进行了表征。红外光谱和电化学研究表明二铁六羰基单元A以二铁五羰基的形式存在于聚合物中。在共聚过程中,增加单体C的含量能显著改变聚合物的形貌和增加其在有机溶剂中的溶解度,同时热稳定性也有所增加。这些聚合物在含有醋酸的DMF介质中催化质子还原的行为表明,通过单体C引入的碱性基团所形成的次级配位环境对二铁中心的催化性能有显著影响。
[FeFe]-氢化酶;聚合物;催化;共聚;“点击”化学
0 引言
当代社会能源危机日益严峻,使得探索和研究可持续、环境友好的新型能源以减轻乃至完全脱离对化石能源的依赖已成为各国科技工作者必须面对的挑战。众所周知,氢能作为未来重要的能源之一,被认为是一种理想的可能取代化石能源的储能物质。因此,如何高效、绿色地制备氢气成为了科学界研究的难点和热点[1]。然而当前人们制备氢气主要采用电解水的方式以及利用贵金属制氢催化剂,前者能源消耗大,后者由于贵金属本身的价格以及催化能力的问题,均限制了氢气的大规模生产和使用,故而研制廉价高效的制氢催化剂就成为突破这一难题的必经之路。另一方面,大自然已经给我们提供了一条可参考的途径氢化酶能够在温和的条件下快速高效地催化产氢。因此,如果能够利用氢化酶产氢机理设计合成催化制氢的模拟体系,将对氢能的发展产生重要的意义。
氢化酶是一种含Fe或Ni金属簇的蛋白质,它广泛存在产甲烷菌、固氮菌、光合成菌、硫还原菌等许多微生物中,具有催化制氢的特殊性质[2]。根据催化活性中心所含金属的不同,氢化酶可以分为三类[3]:[FeFe]-氢化酶、[NiFe]-氢化酶和[Fe]-氢化酶。[FeFe]-氢化酶因其较其它两种酶具有更为高效的催化活性,而受到了最为广泛的关注。自[FeFe]-氢化酶的晶体结构被解析出来之后(活性中心可能的分子结构如Scheme 1所示)[4-5],国内外的科研工作者在化学模拟[FeFe]-氢化酶[2Fe2S]活性中心方面开展了大量的研究工作[3,6-10],极大地增进了人们对[FeFe]-氢化酶的了解。

Scheme 1Schematic view of the H-cluster of the [FeFe]-hydrogenase
另一方面,[FeFe]-氢化酶的金属活性中心处于生物高分子蛋白链的包围之中,这种结构既为金属中心提供了“藏身”之地,也为底物和产物的进出提供了通道,这些蛋白链上的碱性氨基酸残基更为质子的传输提供了“中继站”[11-13]。因此,通过模拟[FeFe] -氢化酶周围的环境,将有助于人类进一步理解和阐明[FeFe]-氢化酶催化制氢的机理,设计合成有效的制氢催化剂。相对于结构模拟研究,目前关于[FeFe] -氢化酶的活性中心周围大分子蛋白的保护以及电子传递的作用进行模拟的报道较少。Darensbourg等[14]用β-环糊精(β-CyD)包裹[FeFe]-氢化酶模型化合物,以此在一定程度上模拟金属中心的生物环境。Artero等[15]将氢化酶的模型化合物修饰至多壁碳纳米管上,利用碳纳米管的优良电子传导性质模拟大分子蛋白在催化制氢的电子传递作用。Pickett小组[16]将含[2Fe3S]单元的模型化合物和[4Fe4S]立方烷同时组装到了电聚合的聚吡咯导电聚合物上,制备了含模拟氢簇的聚合物体系的催化电极。近年来,我们课题组也致力于将高分子化合物与模型化合物相结合并将之用于电极组装的研究。采用的方法有如下3种:含有炔基的二铁模型化合物聚合得到功能聚合物[17]、含炔基的模型化合物与有机叠氮化合物之间的成环聚合[18]、以二铁模型化合物功能化廉价易得的聚氯乙烯(PVC)[19]或聚乙烯吡咯烷酮(PVP)[20],再以自组装或旋转涂膜技术将有关的聚合物组装到电极表面制备催化电极。研究结果表明,这种直接将[FeFe]-氢化酶模型化合物通过聚合反应形成的功能材料,对模型化合物起到一定的保护作用,表现出一定的催化效率,使得受生物启发的[FeFe]-氢化酶模拟研究工作朝着具有实用性的方向推进。
前期我们报道了用二元单体进行聚合的研究[18],其中[FeFe]-氢化酶模型化合物[Fe2(μ-SCH2C≡CH)2(CO)6](A)与二叠氮化合物2,6-(N3CH2)Py(B)聚合得到Poly-Py,而单体A与1,3-二叠氮甲基苯(1,3-(N3CH2)2Ph)反应得到另一种聚合物Poly-Ph。结果发现这两种功能聚合物在溶解性、形貌和电化学性能均存在明显的差别。含有碱性基团的Poly-Py的电化学表现出更好的优越性,聚合物在HBF4中能稳定存在数十小时,起始还原电位比普通的单分子二铁六羰基化合物正移400 mV左右;而另外一种聚合物Poly-Ph不溶于任何有机溶剂。为了进一步探究氢化酶活性中心大分子的蛋白质环境,结合蛋白质含有大量碱性基团的特点,我们在前期工作的基础上[18],设计合成了另一种含有碱性基团的二炔化合物单体C,采用将模型化合物单体A、二叠氮化合物单体B和二炔化合物单体(2-PyCH2)N(CH2C≡CH)2(C)共聚的方法,利用“点击”化学调节聚合物中碱性基团的含量和有机骨架组成,以此考察非模型化合物部分的次级配位环境对聚合物的物理性质和催化性能的影响。
1 实验部分
1.1 实验仪器与基本操作
所有涉及对空气敏感的配体以及金属配合物的合成操作,均采用标准Schlenk无水无氧技术。绝大部分的试剂为市售的AR级化学品,部分试剂购于Alfa Aesar与Sigma-Aldrich公司,实验所用溶剂也预先经过标准的脱水除氧处理。电化学实验采用经典的三电极系统在氩气的气氛下操作完成[21],具体为称取一定量的聚合物加入到0.1 mol·L-1[t-Bu4N] BF4-DMF溶液中,以0.1 V·s-1的速率进行扫描,得到的所有电化学数据均经过二茂铁校正。用于扫描电镜的样品,通过将超声过的聚合物DMF溶液滴加到ITO玻璃电极上风干制备而得。
实验中使用的仪器型号如下:红外光谱仪为Scimitar2000(Varian),核磁共振仪为BrukerAvance DRX(400MHz),元素分析为HeraeusCHN-O-Rapid Elementar Vario MICRO(德国),热重分析为TA SDTQ600,扫描电镜为Quanta200F(FEI,Netherlands),电化学工作站为AutolabPotentiostat30。
1.2 单体及聚合物的合成及表征
1.2.1 化合物A的合成
1.2.1.1 (μ-S2)Fe2(CO)6的合成
在氩气气氛下依次向500 mL的三颈瓶中加入Fe(CO)5(25 mL)和125 mL干燥的甲醇,然后慢慢滴加75 mL已除空气的50%KOH水溶液。室温搅拌30 min后将反应液冷却至0℃,缓慢加入33 g S8,反应液立刻变黑并剧烈放热。试液在0℃下继续搅拌60 min,加入150 mL水、300 mL石油醚后用恒压滴液漏斗加入85 g NH4Cl的150 mL水溶液。撤去冰浴,室温下反应16 h,至有机相呈深红色。分出有机相后再加入200 mL石油醚萃取水相,合并有机相,减压旋干后得深红色固体,利用乙腈进行重结晶,得到红色晶体产物(9.68 g,30.3%)。红外光谱(乙腈,cm-1):ν(CO),2 083(m)2 041(s)2 002(s).熔点:51~52℃.
1.2.1.2 (μ-SCH2CCH)2Fe2CO6的合成
在氩气气氛下将(μ-S2)Fe2(CO)6(0.58 g,1.7 mmol)溶于50 mL干燥的THF,干冰/丙酮浴冷却至-78℃。缓慢滴加LiEt3BH(3.4 mL,1 mol·L-1THF溶液),试液变为墨绿色,搅拌20 min,缓慢滴加3-溴丙炔(0.38 mL,3.4 mmol,80%甲苯溶液),撤去干冰/丙酮浴,室温下反应4 h至试液完全变为红色,减压抽除溶剂,粗产物采用柱色谱分离(洗脱剂:石油醚),可得红色固体化合物A(0.138 g,20%)。红外光谱(二氯甲烷,cm-1):ν(CO),2 077(m)2 043(s)2 004(s)。
1.2.2 化合物B的合成
1.2.2.1 2,6-二溴甲基吡啶(C7H7Br2N)的合成
在氩气气氛下向偶氮异丁氰(AIBN,100.4 mg)和N-溴代丁二酰亚胺(NBS,10.70 g,60.12 mmol)的CCl4(100 mL)溶液中加入2,6-二甲基吡啶(3.5 mL,30 mmol),光照(200 W白炽灯)下搅拌回流2 h。试液变为淡黄色,并有大量的悬浮物形成。反应停止后,过滤除去不溶物,减压旋干溶剂,残留的油状物通过柱色谱进行分离(V乙酸乙酯/V石油醚=1/6),得白色的晶状固体(1.28 g,17%)。熔点:92℃,1H NMR(CDCl3, 295.5 K):4.55(s,CH2),7.38(d,Py,J=7.8 Hz),7.70(q, Py).
1.2.2.2 2,6-叠氮甲基吡啶(C7H7N7)的合成
在氩气气氛下将2,6-二溴甲基吡啶(1.75 g,6.67 mmol)逐滴加入到NaN3(6.50 g,100 mmol)的DMF(50 mL)悬浊液中,并于80℃搅拌反应10 h。反应完全后停止加热,将反应液降至室温,然后往其中加入100 mL蒸馏水,接着用乙醚萃取3次(3×50 mL),合并有机相,用饱和食盐水反萃取有机相3次(3×20 mL),加入适量的无水硫酸镁干燥。过滤除去硫酸镁,减压旋干,残留的无色油状物通过柱色谱进行分离(V乙酸乙酯/V石油醚=1/3),得无色油状化合物B(1.22 g, 96%)。IR(nujol):ν(-N3)2 104 cm-1.1H NMR(CDCl3, 295.5K):4.48(s,4H,CH2),7.30(d,2H,Py,J=7.6 Hz), 7.76(q,2H,Py).
1.2.3 化合物C的合成
在氩气气氛下将3-溴丙炔(1.2 mL,10.00 mmol)缓慢加入至20 mL的氨甲基吡啶(0.5 mL,5.0 mmol)的THF溶液中,快速搅拌下再缓慢加入1.5 mL的Et3N,反应液在室温下搅拌反应18 h。停止反应后过滤除去白色不溶物,减压旋干溶剂,残留的黄色油状物通过柱色谱进行分离(V乙酸乙酯/V石油醚=2/3),得浅黄色的液体化合物C(850 mg,87.4%)。IR(nujol): ν(CCH),3 105 cm-1.1H NMR(CDCl3,295.5K):2.27(s, 2H,CCH),3.47(s,2H,CH2),3.85(s,4H,CH2),7.18 (tri,1H,Ph,J=6.1 Hz),7.44(d,1H,Ph,J=7.8 Hz), 7.66(tri,1H,Ph,J=7.6 Hz),8.56(d,1H,Ph,J=4.5Hz).13C NMR(CDCl3,295.5K):42.3,58.9,73.4,78.6, 122.3,123.3,136.6,149.4,158.1。
1.2.4 聚合物(Polymer-1~Polymer-6)的合成
在氩气气氛下往化合物A([Fe2(μ-SCH2C≡CH)2(CO)6])(0.63 g,1.47 mmol)和化合物C(0.12 g,0.63 mmol)的THF溶液(40 mL)中加入二叠氮化合物B (0.40 g,2.1 mmol)的THF溶液(5 mL),然后加入CuI (17.1 mg,0.09 mmol)的Et3N溶液(0.40 mL)。混合液置于40℃油浴中反应18 h,至试液变为近乎黑色的悬浊液后,将其快速倾倒至急速搅拌的正己烷(1 000 mL)中,持续快速搅拌20 min,然后置于室温下沉降48 h,反应瓶底部析出大量的黑色沉淀。减压抽滤后粗产物依次用THF(250 mL),H2O(100 mL), Et2O-EtOH 1∶1(250 mL)和Et2O(250 mL)洗涤,收集深黄色固体Polymer-1(0.71g,63%),真空干燥后在氩气气氛下进行封存。
聚合物Polymer-2~Polymer-6的合成与上述聚合物Polymer-1的实验操作相同,只是采取了不同的投料比,合成聚合物Polymer-1~Polymer-6的投料比及产物的产率列于表1。
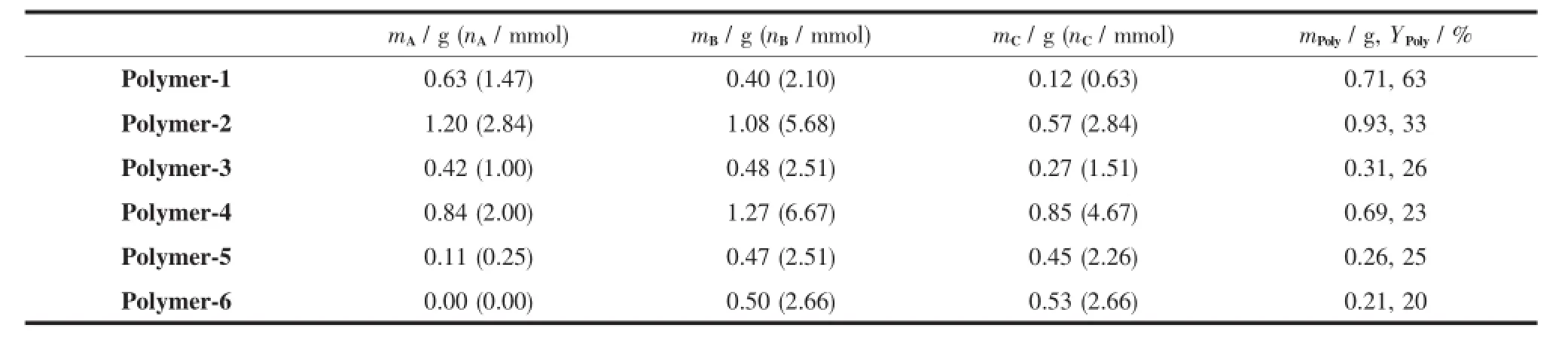
表1 合成聚合物Polymer-1~Polymer-6所需的反应物及产率Table1 Quality of reactants and the yields for the synthesis of Polymer-1~Polymer-6
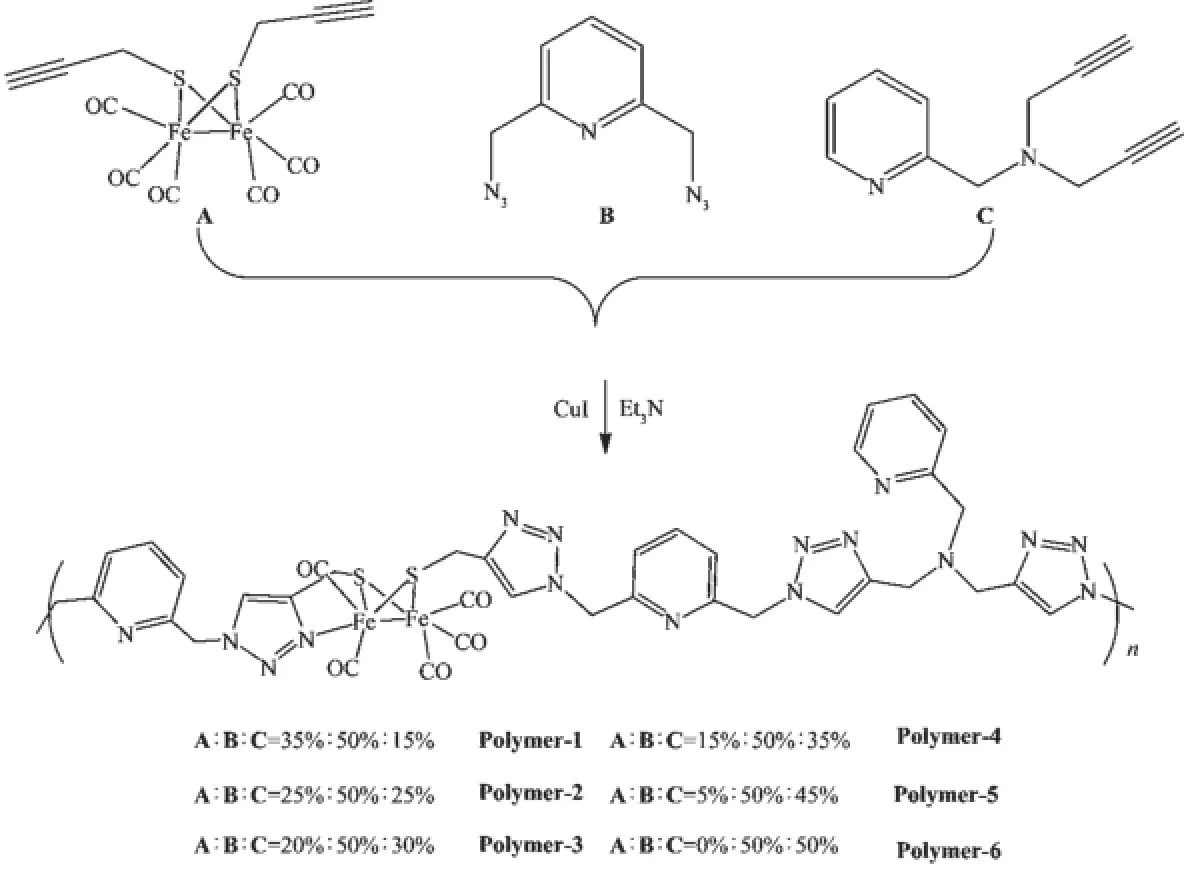
Scheme 2Synthesis of Polymer-1~Polymer-6
2 结果与讨论
2.1 聚合物Polymer-1~Polymer-6的合成
研究表明,在氢化酶催化质子还原过程中,活性中心周边的蛋白环境在质子传递方面起到了重要作用[22-23]。因此,我们在合成的聚合物中引入碱性基团C,以增加聚合物的质子亲和力和亲水性,以此来模拟天然的碱性氨基酸的残基在氢化酶催化过程中所起到的作用。Polymer-1~Polymer-6合成路线及聚合比例如Scheme 2和表1所示。化合物A和B按照文献报道的方法合成[18],化合物C以氨甲基吡啶和3-溴丙炔为原料,在三乙胺的条件下可以高产率地合成得到。为了考察加入的单体C对聚合物性质的影响,我们研究了6种不同的聚合比例(Scheme 2)。为了使6个聚合物具有良好的对比性,所有聚合反应均采用了相同的反应条件:溶剂(THF)、催化剂(CuI)、助剂(Et3N)、反应温度(40℃)以及反应时间(18 h)。
在我们之前报道的聚合体系中,由A与B聚合而得到的聚合物Poly-Py仅能溶于DMF与DMSO[18]。而本文合成的6个聚合物在溶解度方面有很大地改善,不仅能溶于强极性溶剂如CH3OH、CH3CH2OH、DMF与DMSO,而且在中弱极性的溶剂(如THF、DCM)中也有良好的溶解度。这类聚合物溶解性的改善可能得益于聚合体系中引入了含碱性基团单体C,使之在以下2个方面发生了明显的变化:(1)聚合物碱性基团增加,使得聚合物的整体极性明显增强,更容易溶于强极性溶剂中;(2)另一方面,不管是单体B还是C,均含有刚性的吡啶基团,聚合后使得产物的刚性及有机成分增加,导致在中等或弱极性的溶剂中的溶解度变大。
为了验证聚合反应是否按照我们设定的6种投料比进行,我们对Polymer-1~Polymer-5进行了硫元素分析(Polymer-6未引入单体A,故聚合物中不存在硫元素)。从图1可以看出,实际所测硫元素含量均高于基于投料比例的理论值,但总体变化趋势一致。产生这种实验误差的原因可能包括以下两个方面:一是发生聚合反应时含二炔基的[2Fe2S]模型化合物单体A的竞聚率高于单体C;二是单体A在发生聚合反应时存在少量分解的现象,导致铁原子流失而硫原子含量升高从图3可以进一步看出,随着含硫单体A投料的减少,实验值与理论值的误差变小;而投料比比较接近的Polymer-1与Polymer-2、Polymer-3与Polymer-4中硫元素的含量大致相等。
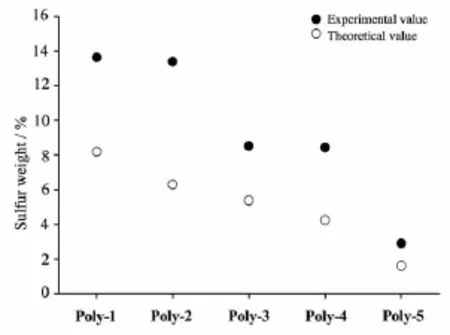
图1 Polymer-1~Polymer-5中实际所测硫元素质量百分数(实心圆)与按投料比计算的理论值(空心圆)的比较Fig.1 Comparison of the measured mass percentages (solid)of sulfur with those calculated(circle)in Polymer-1~Polymer-5 based on the stoichiometric ratios of the reactions
纵观这些聚合反应的产率,除了Polymer-1的产率高于50%之外,其余聚合物的产率均在20%~30%之间,这也进一步说明由于单体A的竞聚率高于单体C,单体A优先发生聚合反应,导致聚合的组成并不与理论一致。而当单体A的投料减少时,单体C参与聚合的机率增加,产物中各种组分的比例趋近于投料比,故实验值与理论值之间的差异变小。
2.2 Polymer-1~Polymer-6的表征
如图2所示,Polymer-1~Polymer-5这5个含有羰基铁单元的聚合物的红外模式与我们之前报道的2种聚合物(Poly-Py和Poly-Ph)一致[18],均是文献中报道的五羰基化合物的红外模式[24-28]。这就意味着在共聚过程中,单体A的1个羰基被其附近的1个三唑环所取代[18]。红外数据表明:通过这种共聚的方法,能够可控地将不同含量的[FeFe]-氢化酶[2Fe2S]模型化合物枝接到聚合物上,并且二铁单元的配位模式和之前研究的由2种单体形成的聚合物(Poly-Py和Poly-Ph)相同[18],即由“点击”反应得到的三唑环上的1个N原子与附近的二铁六羰基单元中的1个Fe原子进行配位,从而形成二铁五羰基单元的体系。
考虑到聚合物(Polymer-1~Polymer-5)中均含有大量碱性基团,对质子有较强的结合能力,我们进一步考察了这几个聚合物在醋酸条件下的质子化情况,并用红外光谱检测其质子化前后的变化。图3~5为聚合物Polymer-1~Polymer-5质子化前后的红外对比图。不难看出,质子化前后红外模式没有发生明显地变化,只是在吸收峰强度上有微小地减弱。这些实验结果表明:聚合物Polymer-1~Polymer-5在醋酸的存在条件下具有良好的稳定性,二铁单元的配位模式也没有发生改变,说明这类聚合物体系对二铁中心可以起到较好的保护作用。

图2 Polymer-1~Polymer-6的红外谱图(羰基部分)Fig.2 IR spectra of Polymer-1~Polymer-6(carbonyl region)
我们进一步对Polymer-1~Polymer-6进行了热重分析。从图6中可以看出,不含铁羰基单元的Polymer-6的热重曲线较含有铁羰基的Polymer-1~ Polymer-5的失重情况表现出明显的不同:(1)从364~557 K,Polymer-6的失重不到8%,而Polymer-1~Polymer-5的失重均接近20%(其中含铁羰基单元多的Polymer-1超过20%);(2)在730~1 100 K阶段,Polymer-6的失重率约为20%,而Polymer-1~Polymer-5的失重率不足10%(其中Polymer-1的失重率最小)。结合制备聚合物Polymer-1~Polymer-6的投料比以及热重图,我们不难得出:(1)聚合之后铁羰基单元的分解主要集中在364~557 K阶段;(2)在高温阶段(730~1 100 K),铁硫含量越高,失重率越小。
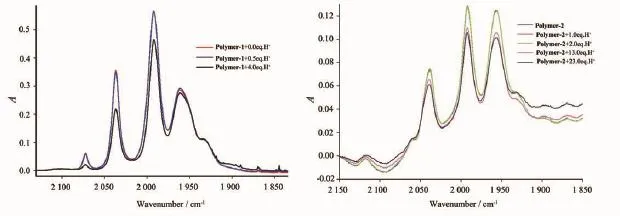
图3 Polymer-1和Polymer-2的加醋酸前后红外对比图Fig.3 IR spectral changes of Polymer-1 and Polymer-2 before and after the addition of acetic acid
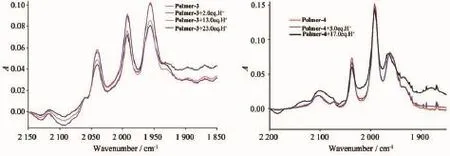
图4 Polymer-3和Polymer-4的加醋酸前后红外对比图Fig.4 IR spectral changes of Polymer-3 and Polymer-4 before and after the addition of acetic acid

图5 聚合物Polymer-5在加醋酸前后的红外对比图Fig.5 IR spectral changes of Polymer-5 before and after the addition of acetic acid
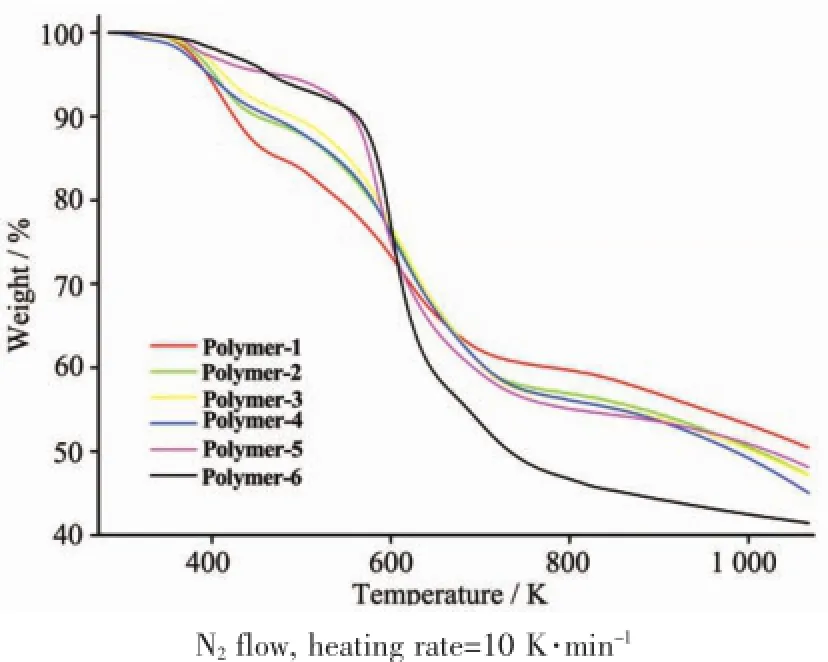
图6 聚合物Polymer-1~Polymer-6的热重分析(TGA)图Fig.6 TGA diagrams of Polymer-1~Polymer-6
为进一步了解聚合物中碱性基团增加对聚合物具体形貌的影响,我们对其进行了扫描电镜的测试,其结果如图7所示。
从扫描电镜图(图7)可以清楚的看出,随着碱性基团的增加即从Polymer-1~Polymer-6,聚合物的球状物粒径逐渐增大,逐渐从粒状(Polymer-1)过渡到球状(Polymer-2~Polymer-4)再到片状(Polymer-5和Polymer-6)。聚合物形貌的变化可以从聚合反应的投料比例加以解释:从Polymer-1~Polymer-6,含碱性基团单体C的含量逐渐增加,粒子之间结合能力增强,使得聚集状态从小到大发生变化。
2.3 聚合物Polymer-1~Polymer-6电化学性质研究前期研究结果表明,含铁羰基模型化合物的聚合物材料在催化质子还原时的电化学行为与小分子活性中心存在着较大的区别[18],为了进一步对聚合物的催化性质进行探索,我们研究了Polymer-1~Polymer-6在DMF介质中的循环伏安电化学行为(图8)。

图7 Polymer-1~Polymer-6的扫描电镜图(放大50 000倍)Fig.7 SEM images of Polymer-1~Polymer-6(magnified by 50 000 times)
如图8所示,在没有加酸的情况下,这些聚合物在-1.5 V左右(六羰基铁单元的还原电位)和-1.75 V左右(五羰基铁单元的还原电位)的还原峰均清晰可见,与文献报道的类似化合物有相似的电化学行为[26-28]。只是由于二铁含量的不同,-1.5 V处的还原峰会有相对不同的峰高值,并随着铁羰基单元的含量的减少而减弱。在这类由“点击”化学反应所制备的聚合物中,绝大多数的二铁单元都分布在聚合物的大分子结构中,且都已被取代形成五羰基的单元,只有很少量的处于端基位置的二铁单元未发生取代,依然保持着六羰基的模式。然而,当整个聚合物体系含铁羰基单元量少时,例如Polymer-5(二铁单元的含量仅5%),处于端基的二铁单元的量大大降低,因此在该聚合物中几乎看不到六羰基铁单元的还原峰。

图8 Polymer-1~Polymer-6在[t-Bu4N]BF4/DMF中加入冰醋酸前后的循环伏安图Fig.8 Cyclic voltammograms of Polymer-1~Polymer-6 before and after the addition of acetic acid in[t-Bu4N]BF4/DMF
在外加醋酸的条件下,这些聚合物的电化学行为远比单分子化合物复杂。如图8所示,随着醋酸的加入,在约-1.2 V处产生一个新的还原峰,并且在约-0.6 V处出现了一组可逆的氧化还原峰。这2个还原峰都是在加入质子之后产生的,除Polymer-1外,其相对强度会随着二铁单元含量的减少而逐渐减弱。由此可知,-1.2 V处和-0.6 V处所产生的新还原峰不但与二铁单元的含量有关,还与质子相关。对于聚合物Polymer-1来说,其循环伏安图几乎看不到-0.6 V处的还原峰,这可能由于该聚合物中二铁的含量比其他聚合物都高,因此在循环伏安扫描时,由于二铁单元相关还原峰都比其他聚合物要强,从而掩盖了在-0.6 V处的还原峰。另一方面,这些聚合物在加酸的情况下,各还原峰峰高均随酸的加入而增加,但到一定浓度之后增加的幅度就逐渐减慢,有些甚至不再增强,表明聚合物接受质子的能力有限,一定程度之后就会达到饱和,这与我们之前报道的聚合物Poly-Py和Poly-Ph相似[18]。
我们进一步考察了这些聚合物中由二铁五羰基单元产生的还原峰(-1.75 V附近)峰电流强度随加入的醋酸当量的变化情况。如图9所示,随着醋酸的加入,imol(单位物质的量浓度的还原电流强度)逐渐增加,但到一定浓度之后增加的幅度就逐渐减慢,直至饱和。结合之前的硫元素分析结果,聚合物体系中实际的硫含量偏高,说明聚合物引入的二铁模型化合物比实际的投料比要多,而含氮碱性基团的含量比实际的投料比小,导致聚合物缓冲质子的能力下降,催化饱和更容易出现。
显而易见,这类聚合物在加酸之后的电化学与普通单分子二铁六羰基和五羰基化合物有着明显的差别[24-32]。文献报道表明,当同一个[2Fe2S]模型化合物的羰基被不同的取代基团取代后,其产物的质子化行为会有显著的差异[33]。由此可见,模型化合物的配位环境对模型化合物的质子化起着举足重轻的作用。Polymer-1~Polymer-5聚合物体系中,大量的叔胺基团、吡啶环、三唑环等碱性基团构建了丰富的次级配位环境,导致了如上所述的电化学特征。
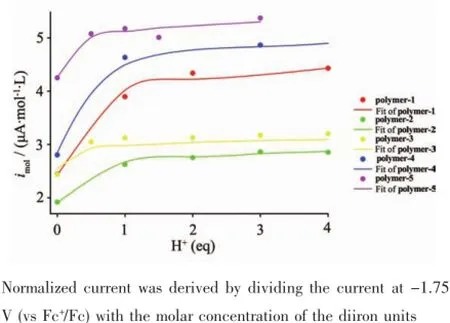
图9 Polymer-1~Polymer-5在-1.75 V(vs Fc+/Fc)处的归一化还原电流(imol)随醋酸浓度的变化Fig.9 Normalized catalytic current against the concentration of acetic acid for Polymer-1~Polymer-5
3 结论
采用“点击”反应,将含二炔基的[2Fe2S]模型化合物A与双叠氮单体B和含碱基的双炔单体C进行不同比例的共聚,制备得到了6种含不同碱基量的聚合物Polymer-1~Polymer-6,并对其进行了红外、硫元素分析、热重、扫描电镜和电化学的表征。实验结果表明:不同含量的碱性基团对含[2Fe2S]活性中心的聚合物的稳定性和催化性能均表现出显著的影响。这种由多个单体改变投料比例进行共聚的方式,不但极大地改善了聚合物在有机溶剂中的溶解性,而且在[2Fe2S]催化活性中心周围构建了富含碱基的“保护罩”,既稳定了活性中心又提高了催化效率,为[FeFe]-氢化酶二铁活性中心催化反应环境的研究提供有益的参考。
[1]Baños R,Manzano-Agugliaro F,Montoya F G,et al.Renewable Sustainable Energ Rev.,2011,15(4):1753-1766
[2]Adams M W W,Mortenson L E,Chen J S.BBA-Bioenergetics, 1980,594(2/3):105-176
[3]Tard C,Pickett C J.Chem.Rev.,2009,109(6):2245-2274
[4]Peters J W,Lanzilotta W N,Lemon B J,et al.Science, 1998,282(5395):1853-1858
[5]Nicolet Y,Piras C,Legrand P,et al.Struct.Fold.Des., 1999,7(1):13-23
[6]Darensbourg M Y,Lyon E J,Smee J J.Coord.Chem.Rev., 2000,206:533-561
[7]Best S P.Coord.Chem.Rev.,2005,249(15/16):1536-1554
[8]Capon J F,Gloaguen F,Schollhammer P,et al.Coord.Chem. Rev.,2005,249(15/16):1664-1676
[9]Liu X M,Ibrahim S K,Tard C,et al.Coord.Chem.Rev., 2005,249(15/16):1641-1652
[10]Fontecilla-Camps J C,Volbeda A,Cavazza C,et al.Chem. Rev.,2007,107(10):4273-4303
[11]Pilet E,Nicolet Y,Mathevon G,et al.FEBS Lett.,2009, 583:506-511
[12]Ginovska-Pangovska B,Ho M H,Linehan J C,et al.BBABioenergetics,2014,1837(1):131-138
[13]Shepard E M,Mus F,Betz J N,et al.Biochemistry,2014,53 (25):4090-4104
[14]Singleton M L,Reibenspies J H,Darensbourg M Y.J.Am. Chem.Soc.,2010,132(26):8870-8871
[15]Le Goff A,Artero V,Jousselme B,et al.Science,2009,326 (5958):1384-1387
[16]Ibrahim S,Woi P M,Alias Y,et al.Chem.Commun.,2010, 46(43):8189-8191
[17]Ru X,Zeng X,Li Z,et al.J.Polym.Sci.,Part A:Polym. Chem.,2010,48(11):2410-2417
[18]Zhan C,Wang X,Wei Z,et al.Dalton Trans.,2010,39(46): 11255-11262
[19]Wang L J,Xiao Z Y,Ru X,et al.RSC Adv.,2011,1(7): 1211-1219
[20]Xu E,Xiao Z,Liu H,et al.RSC Adv.,2012,2(27):10171-10174
[21]Li Z,Zeng X,Niu Z,et al.Electrochim.Acta,2009,54(13): 3638-3644
[22]Vincent K A,Parkin A,Armstrong F A.Chem.Rev.,2007,107 (10):4366-4413
[23]Roy S,Shinde S,Hamilton G A,et al.Eur.J.Inorg.Chem., 2011(7):1050-1055
[24]Xiao Z Y,Xu F F,Long L,et al.J.Organomet.Chem., 2010,695(5):721-729
[25]Xu F,Tard C,Wang X,et al.Chem.Commun.,2008,(5): 606-608
[26]Capon J F,El Hassnaoui S,Gloaguen F,et al.Organometallics, 2005,24(9):2020-2022
[27]Razavet M,Davies S C,Hughes D L,et al.Dalton Trans., 2003(4):586-595
[28]Gloaguen F,Lawrence J D,Schmidt M,et al.J.Am.Chem. Soc.,2001,123(50):12518-12527
[29]Xiao Z,Wei Z,Long L,et al.Dalton Trans.,2011,40(16): 4291-4299
[30]Zeng X H,Li Z M,Xiao Z Y,et al.Electrochem.Commun., 2010,12(3):342-345
[31]Surawatanawong P,Tye J W,Darensbourg M Y,et al.Dalton Trans.,2010,39(12):3093-3104
[32]Ezzaher S,Capon J F,Dumontet N,et al.J.Electroanal. Chem.,2009,626(1/2):161-170
[33]Ezzaher S,Gogoll A,Bruhn C,et al.Chem.Commun., 2010,46(31):5775-5777
Synthesis and Characterization of[FeFe]-Hydrogenase Model Complex Functionalized Polymers Based on Different Content of Alkaline Group
LIANG Shao-Wei1ZHONG Wei1ZHAN Cai-Xia2ZHAO Jia2LI Wen-Qiang2YE Ping1WANG Hong-Dan1SHEN Jie1FAN Lu1XIAO Zhi-Yin1LIU Xiao-Ming*,1,2
(1College of Biological,Chemical Sciences and Engineering,Jiaxing University,Jiaxing,Zhejiang 314001,China)
(2Department of Chemistry,Nanchang University,Nanchang,330031,China)
Co-polymerization of[2Fe2S]model complex[Fe2(μ-SCH2C≡CH)2(CO)6](A),diazide 2,6-(N3CH2)Py(B, Py=pyridine)and diyne compound(2-PyCH2)N(CH2C≡CH)2(C,Py=pyridine)led to the formation of six functionalized Polymers Polymer-1~Polymer-6 via“click reaction”by varying the ratio of the three components. These Polymers were characterized by IR spectroscopy,sulfur elemental analysis,thermal gravimetric analysis, scanning electron microscopy and electrochemistry.IR spectroscopic data and electrochemical investigations suggested that diiron hexacarbonyl complex A exists as diiron pentacarbonyl unit in these Polymers.In the copolymerizing process,increasing the content of monomer C can significantly change the morphologies of these Polymers and improve their solubility in organic solvents and thermal stability.Electrochemistry of these Polymers in acetic acid-DMF indicated that the secondary coordination atmosphere established by introducing alkaline group provided by the monomer C can significantly enhance the catalytic performance of the diiron units in these Polymers.
[FeFe]-hydrogenase;polymer;catalysis;Co-polymerization;“click”chemistry
O614.81+1;O633.2
A
1001-4861(2015)01-0087-10
10.11862/CJIC.2015.011
2014-07-08。收修改稿日期:2014-09-18。
国家级大学生创新训练计划项目(No.201310354021)和国家自然科学基金(No.21171073)资助项目。*
。E-mail:xiaoming.liu@mail.zjxu.edu.cn,Tel/Fax:0573-83643937
猜你喜欢
玻璃(2022年1期)2022-02-23
陶瓷学报(2021年5期)2021-11-22
水泵技术(2021年4期)2021-01-22
新课程(下)(2018年4期)2018-03-26
计算机系统应用(2017年5期)2017-06-07
山东工业技术(2016年15期)2016-12-01
科技传播(2016年17期)2016-10-10
橡胶工业(2015年2期)2015-07-29
中国塑料(2014年12期)2014-10-17
应用化工(2014年1期)2014-08-16
