
炭载Pd-Fe合金催化剂的制备及电催化氧还原活性
2015-06-01 10:30王彦恩刘书静唐亚文陆天虹
无机化学学报 2015年1期
王彦恩 曹 爽*, 刘书静 冯 涛 刘 宁 唐亚文 陆天虹
(1河北农业大学理学院,河北农业大学现代科技学院,保定071000)
(2南京师范大学化学与材料科学学院,南京210097)
炭载Pd-Fe合金催化剂的制备及电催化氧还原活性
王彦恩1曹 爽*,1刘书静1冯 涛1刘 宁1唐亚文*,2陆天虹2
(1河北农业大学理学院,河北农业大学现代科技学院,保定071000)
(2南京师范大学化学与材料科学学院,南京210097)
研究了用NH4Cl作配位剂的配位还原法来制备的Pd-Fe/C催化剂,发现由于NH4Cl能与Pd形成配合物,使PdCl2的还原电位负移,与FeCl3的还原电位接近,从而在低温下制备得到了高合金化程度的Pd-Fe/C催化剂。XPS表征结果表明:Pd与Fe形成合金后,Pd的电荷密度的减少,增加了Pd0的含量。因此,得到的Pd-Fe/C催化剂对氧还原的电催化活性比用相同方法制得的Pd/C催化剂高,而且该催化剂对甲醇氧化没有电催化活性。
催化剂;配位还原法;还原电位;合金化程度;氧还原
0 前言
DMFC由于甲醇来源丰富、价格低廉、贮存与携带方便,且能量密度高等特点,有良好的应用前景[1]。Pt/C催化剂是目前DMFC中主要使用的阴极电催化剂,但Pt对甲醇氧化有很好的电催化活性,甲醇渗透至阴极在阴极形成“混合电位,且甲醇氧化产物CO对催化剂有毒化作用而使Pt/C阴极催化剂对氧还原的电催化性能降低,加上Pt资源有限[2],因此,研究对氧还原有高电催化活性和价格低廉的抗甲醇的DMFC的阴极催化剂,无论在基础研究领域还是对DMFC的商品化开发均具有极其重要的意义[3-5]。
在众多催化剂中,Pd催化剂对氧还原有好的电催化性能,但对甲醇氧化无电催化活性[6]。而且与Pt相比,Pd来源较丰富,价格便宜,因此,Pd作为DMFC中的阴极催化剂引起了研究者广泛兴趣。但Pd催化剂对氧还原的电催化活性要比Pt催化剂低,因此,许多研究都集中在Pd基复合催化剂方面。载体上高度分散的Pd-Fe纳米颗粒,已经引起了人们的广泛重视[7-25]。
这些有关Pd与过渡金属Fe形成合金化的催化剂,首先用传统化学方法还原,然后500~900℃高温热处理促进合金形成[7,21-22,26-27]。但是,高温热处理,会导致颗粒聚集,降低催化剂的表面积及催化活性。相比较而言,可以通过直接热分解有机金属前驱体获得Pd-Fe合金纳米颗粒[10,24,28],但是此方法材料昂贵,限制了它的广泛应用。
本工作用配位还原法在低温下合成合金化程度较高、粒径较小的Pd-Fe/C催化剂,与Pd/C催化剂相比,Pd-Fe/C催化剂对氧还原的起始电位提高了将近90 mV。
1 实验部分
1.1 试剂与仪器
Vulcan XC-72活性炭(美国Cabot公司)和Nafion(全氟聚苯乙烯磺酸)溶液(5wt%,美国Sigma-Aldrich化学公司产品)均直接使用;氯化钯,上海试剂一厂,分析纯;其余试剂均为分析纯试剂,所有溶液均用三次蒸馏水配制。
电化学测量用CHI600电化学分析仪(美国CHI仪器公司)和常用的三电极电化学池进行。工作电极为瑞士万通的Auto-Lap旋转圆盘电极,其基体为玻碳电极(直径3 mm),用Pt片作为对电极,Ag/AgCl为参比电极,文中所引用的电位均相对于Ag/AgCl电极。用高分辨电子显微镜(HRTEM,JEM-2100,加速电压200 kV)表征了产物的形貌和微观结构。电感耦合等离子体原子发射光谱(ICP)用JY/T 015-1996感耦等离子体原子发射光谱仪。X-射线衍射(XRD)测量用D/max-rC型转靶X射线衍射仪(日本理学公司)进行,靶电压40 kV,靶电流100 mA,Cu Kα射线源为光源(λ=0.154 18 nm)。紫外-可见(UV-Vis)吸收光谱测量用德国Perkin-Elmer公司的Lambda17型UV-Vis吸收光谱仪进行。
1.2 催化剂的制备
将4.78 mmol XC-72活性炭加入3.13 mL 0.450 4 mol·L-1的PdCl2水溶液,超声10 min,得活性炭和PdCl2的悬浊液。将1.50 mmol NH4Cl和2.02 mmol H3BO3溶解在10 mL水中,然后加入活性炭和PdCl2悬浊液中,搅拌均匀后再加入3 mL 0.015 7 mol·L-1FeCl3,继续超声1 h,得到混合物悬浊液,将1.06 mmol NaBH4加入10 mL水中,搅拌、90℃水浴中,将上述悬浊液缓慢滴加到NaBH4水溶液中,继续搅拌1 h,使PdCl2和FeCl3与还原剂NaBH4完全反应后,用三次蒸馏水洗多次,最后,在真空条件下55℃干燥,即制得含质量分数为20% Pd的Pd-Fe/C催化剂。
Pd/C催化剂的制备方法同上,只是不加FeCl3。
1.3 电化学测试
工作电极的制备:玻碳电极在使用前依次用6#金相砂纸、0.3和0.05 μm的Al2O3抛光粉抛光至镜面后,在三次蒸馏水中超声洗涤后备用。然后将2.5 mg Pd/C或Pd-Fe/C催化剂与0.4 mL水、0.3 mL乙醇和0.05 mL Nafion溶液配制成悬浮液,超声分散30 min,移取3 μL悬浮液至电极表面,50℃干燥后得工作电极。其Pd载量为28 μg·cm-2。
在进行电化学测量时,电解液为0.5 mol·L-1HClO4溶液,溶液温度控制在30±1℃。线性扫描时,工作电极转速:2 000 r·min-1,电位扫描速率:5 mV· s-1。测量前,先通高纯氮20 min,以除去溶液中的氧,在进行氧还原测量前,通氧20 min,以使溶液中含饱和氧。
2 结果与讨论
2.1 催化剂结构与制备机理表征
图1为PdCl2溶液和PdCl2+NH4Cl+H3BO3溶液的UV-Vis吸收光谱。由图可知,PdCl2水溶液在419,237和207 nm处有3个特征吸收峰(曲线a)。PdCl2溶液中加入NH4Cl+H3BO3后,PdCl2的特征吸收峰消失(曲线b),而在294 nm处出现一个新的特征吸收峰。这是由于NH4Cl在H3BO3缓冲溶液中主要以NH3·H2O形式存在,NH3·H2O与Pd2+能形成[Pd(NH3)4]2+配离子,因此,PdCl2的特征吸收峰消失,而出现[Pd(NH3)4]2+的特征吸收峰。这证明了Pd2+在这种体系中能形成配合物。
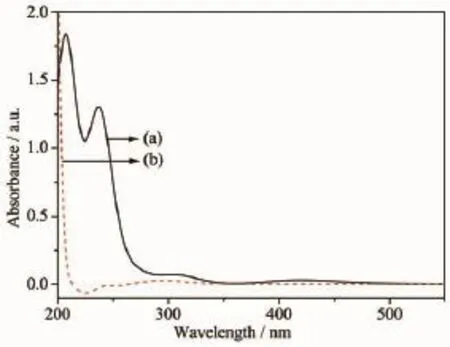
图1 (a)PdCl2溶液和(b)PdCl2+NH4Cl+H3BO3溶液的UV-Vis吸收光谱Fig.1 UV-Vis absorption spectra of PdCl2in(a)without NH4Cl and H3BO3and(b)with NH4Cl and H3BO3solution
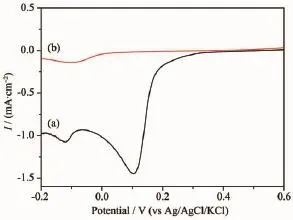
图2 玻碳电极在含(a)0.005 5 mol·L-1PdCl2和(b)0.005 5 mol·L-1PdCl2+NH4Cl+H3BO3的0.05 mol·L-1KCl溶液中的线性扫描伏安曲线Fig.2 Linear sweeping voltammograms of(a)0.005 5 mol· L-1PdCl2and(b)0.005 5 mol·L-1PdCl2+NH4Cl+ H3BO3at the glassy carbon electrode(0.05 mol·L-1KCl solution)
图2 为玻碳电极在含0.005 5 mol·L-1PdCl2和 0.005 5 mol·L-1PdCl2+NH4Cl+H3BO3的0.05 mol·L-1KCl水溶液的线性扫描伏安曲线。由图可见,PdCl2在纯水溶液中的起始还原电位位于0.2 V附近(曲线a),而在含NH4Cl+H3BO3后起始还原电位位于0 V附近(曲线b),比PdCl2纯水溶液中负移了300 mV左右。而FeCl3在纯水溶液中和在含NH4Cl+H3BO3溶液中的起始还原电位都位于-0.1 V附近。PdCl2起始还原电位的负移,是由于PdCl2与NH3·H2O形成配合物引起的。由于PdCl2和FeCl3起始还原电位差变小,这意味着在NH4Cl和H3BO3混合溶液中,Pd和Fe基本上可在相同的电位下还原得到,因此,在NH4Cl和H3BO3混合溶液中,当用还原剂还原PdCl2和FeCl3时,Pd和Fe基本上可同时还原出来,成核和增长速率也基本相同。因此,Fe较容易进入对方晶格形成合金,使制得的Pd-Fe/C催化剂中Pd-Fe粒子的合金化程度较高。
图3为Pd/C和Pd-Fe/C催化剂的EDS谱。其中2种催化剂中Pd的质量百分数分别为19.96%和20.03%,表明加入的Pd和Fe几乎完全被还原出来。Pd-Fe/C催化剂EDS谱中Pd与Fe的原子比为接近3∶1,与理论值很接近,表明Pd和Fe几乎被完全还原出来,并且图谱中没有发现B的残留。通过电感耦合等离子体测试制备的Pd/C和Pd-Fe/C催化剂中Pd和Fe的实际含量,测试结果中Pd约为19.89和19.19%,Fe约为3.3%,Pd与Fe的原子比为接近3∶1。表明Pd和Fe几乎被完全还原出来,与EDS测试结果基本一致。

图3 (a)Pd/C和(b)Pd-Fe/C催化剂的EDS谱Fig.3 EDS spectrum of the(a)Pd/C and(b)Pd-Fe/C catalyst

图3 (a)Pd/C和(b)Pd-Fe/C催化剂的EDS谱Fig.3 EDS spectrum of the(a)Pd/C and(b)Pd-Fe/C catalyst
图4 为Pd/C和Pd-Fe/C催化剂的XRD图。在Pd/C催化剂的XRD图中(曲线a),可观察到在2θ为24.7°处碳(002)晶面的衍射峰。其他衍射峰的2θ值为39.68°,45.42°,67.10°和80.42°,它们分别相应于具有面心立方结构的Pd晶体的Pd(111)、Pd (200)、Pd(220)和Pd(311)的晶面衍射峰,表明Pd/C催化剂中的Pd粒子以面心立方结构形式存在。在Pd-Fe/C催化剂的XRD图中(曲线b),Pd衍射峰峰位比Pd/C催化剂的明显正移,这一方面表明Pd-Fe/ C催化剂中的Pd-Fe粒子也具有面心立方晶体结构,另一方面,表明有部分Fe已进入Pd的晶格,与Pd形成合金,因为Fe的原子半径要比Pd小,因此,Fe进入Pd晶格后,使Pd衍射峰的2θ值变大,发生晶格收缩。另外,在Pd-Fe/C催化剂的XRD图中没有观察到Fe或Fe2O3的特征峰,表明没有进入Pd晶格的Fe或Fe2O3是以无定形的形式存在。由XRD图计算得的2种催化剂的结构数据列于表1。
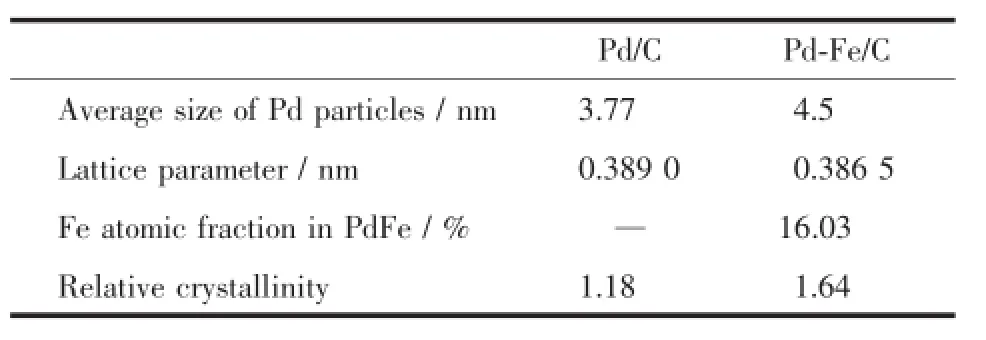
表1 Pd/C及Pd-Fe/C催化剂的晶体结构参数Table1 Structure parameters of(a)Pd-Fe/C and(b) Pd-Fe/C catalysts
由图4数据通过高斯拟合的方法[29],由Pd(220)晶面衍射峰,利用公式(1),计算得Pd-Fe/C催化剂中Pd-Fe粒子的晶格常数是0.386 5 nm,这与用高温500℃热处理制备的合金Pd3-Fe1/C催化剂晶格常数很接近,根据Vegard定律[30],利用公式(2),计算得Pd-Fe合金中与Pd形成合金的Fe的原子分数(相对于Pd)是16.03%,而Pd-Fe/C催化剂的EDS谱图中得到Pd和Fe的原子比为3∶1,即Fe的原子分数为25%,这说明在Pd-Fe/C催化剂中存在着2种形态的Fe:一种为合金态,另一种为非合金态。而用普通NaBH4还原方法制备得Pd-Fe催化剂中,与Pd形成合金的Fe的原子分数(xFe=3.7%)[16],以上结果说明配位还原法可以制备高合金化的Pd-Fe/C催化剂,见表2。

式中:α为Pd-Fe合金的晶格常数,αo为纯Pd晶格常数(0.389 0 nm),κ为常数(0.0156 nm),xFe为与Pd形成合金的Fe的原子分数,λ为射线源的入射波长(0.154 2 nm),θ为衍射角。
如图5所示,用透射电镜(高倍HRTEM)对制备的Pd/C和Pd-Fe/C催化剂进行了表征,从图5可以看到,催化剂中Pd-Fe粒子和Pd粒子良好的分散在碳载体上,并且尺寸分布集中,平均粒径5 nm。HRTEM的研究显示出Pd-Fe金属纳米粒子为均一的单相合金结构(图5b)。

图5 (a)Pd/C和(b)Pd-Fe/C催化剂的HRTEM图Fig.5 HRTEM images of(a)Pd/C and(b)Pd-Fe/C catalysts
图6 为Pd/C、Pd-Fe/C催化剂中Pd3d的XPS谱。在每个催化剂的Pd3d谱中都有2个谱峰,位于336.0和341.0 eV左右,分别对应于Pd3d5/2和Pd3d3/2的结合能[31-33]。由图可知,Pd-Fe/C催化剂的Pd3d5/2结合能在335.8 eV,而Pd/C催化剂中Pd3d5/2谱峰出现在335.4 eV,说明Pd-Fe/C催化剂中的Pd3d5/2电子结合能比Pd/C高0.43 eV,表明了Pd-Fe/C催化剂中Pd的电荷密度的减少,这是由于Fe的电负性强,在Pd-Fe合金中,Pd的电子部分转移到Fe上。Osaka等[34]也报道过相似的结论。这可能就是Pd-Fe/C催化剂对氧还原的电催化性能高于Pd/C催化剂的原因。

表2 Pd-Fe/C催化剂的组成Table2 Composition of Pd-Fe/C catalyst

图6 (a)Pd/C,(b)Pd-Fe/C催化剂Pd3d的XPS谱Fig.6 XPS spectra of(a)the Pd/C,(b)Pd-Fe/C-catalysts in the Pd3d region
通过高斯拟合,把每个Pd3d5/2能谱峰分成2个对应于335.5和337.5 eV的谱峰,它们对应Pd0和PdⅡ的结合能[35]。而不同价态的Pd的含量通过高斯拟合峰的积分面积得到。Pd/C、Pd-Fe/C催化剂中的不同价态的Pd的含量和结合能列于表3中。从表3中可以看出Pd/C催化剂中Pd0的含量仅为51.1%,远小于Pd-Fe/C催化剂中的Pd0的含量。许多研究已经证明Pd比Pt更易氧化[36-37],Pd作为电催化剂稳定性不高。然而Fe的加入,有利于Pd抗氧化性能的增强,因此,Pd与Fe形成合金后,增加了Pd0的含量而提高了Pd对氧还原的电催化活性。
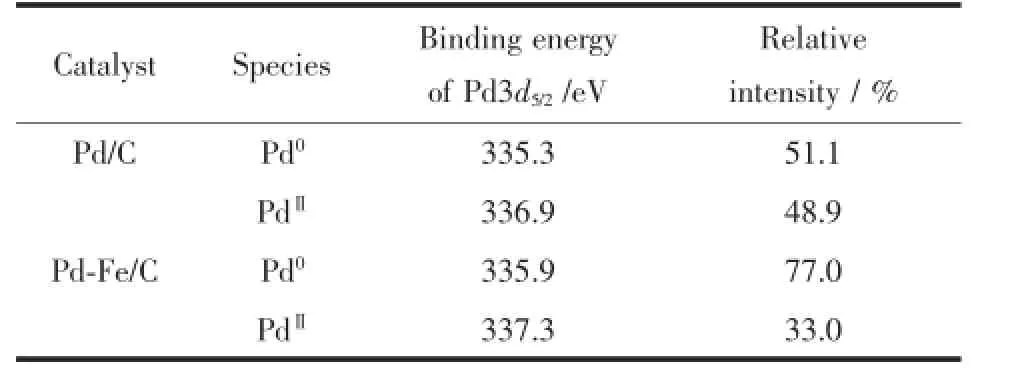
表3 不同形态的Pd的含量及其结合能Table3 Binding energies and relative intensities of Pd0and PdⅡ
2.2 Pd-Fe/C催化剂对氧还原的电催化性能
图7为不同催化剂电极在氧气饱和的0.5 mol· L-1HClO4溶液中的线性扫描伏安曲线。由图可见,在Pd/C、Pd-Fe/C催化剂电极上,氧还原的起始还原电位分别为0.50 V和0.59 V,Pd-Fe/C催化剂对氧还原的起始还原电位要比Pd/C催化剂正90 mV,表明Pd-Fe/C催化剂对氧还原的电催化性能要好于Pd/C催化剂。由TEM表征得Pd-Fe/C催化剂与Pd/ C催化剂中Pd粒子的粒径相近,因此,Pd-Fe/C催化剂对氧还原的电催化活性高于Pd/C催化剂的原因不是Pd粒子结构引起的,Fe与Pd之间合金化形成极大的提高了对氧还原的电催化活性。
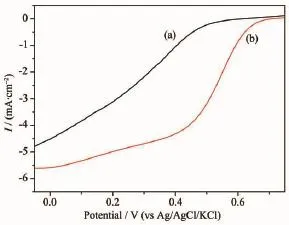
图7 (a)Pd/C催化剂和(b)Pd-Fe/C催化剂电极在含饱和氧的0.5 mol·L-1HClO4溶液中的线性扫描伏安曲线Fig.6 Linear sweep voltammograms of(a)the Pd/C and (b)Pd-Fe/C catalyst electrodes in the 0.5 mol·L-1HClO4solution with saturated oxygen
图8 是Pd-Fe/C催化剂电极在氧气饱和的0.5 mol·L-1H2SO4溶液(a曲线)和含甲醇的0.5 mol·L-1H2SO4溶液(b曲线)中的线性扫描曲线。实验表明,当电解液中有甲醇或没有甲醇时,Pd-Fe/C催化剂在含饱和氧的HClO4溶液中的线性扫描伏安曲线基本一样,而且没有观察到甲醇的氧化峰,表明Pd-Fe/C催化剂对甲醇氧化没有电催化活性,因此,Pd-Fe/C催化剂是一种很好的DMFC的阴极催化剂。

图8 Pd-Fe/C催化剂电极在含(a)非甲醇和(b)甲醇的氧气饱和的0.5 mol·L-1HClO4溶液中的线性扫描曲线Fig.8 Linear sweep voltammograms ofthe Pd-Fe/C catalyst electrodes in the 0.5 mol·L-1HClO4solution(a)without MeOH and(b)in the prensence of MeOH with saturated oxygen
3 结论
由于NH4Cl能与Pd形成配合物,使PdCl2还原电位负移,当用还原剂还原PdCl2和FeCl3时,由于PdCl2和FeCl3起始还原电位差变小,则Pd和Fe成核及增长速率也基本相同,Fe较容易进入Pd晶格,从而在低温下制备了合金化程度较高的Pd-Fe/C催化剂。与Pd形成合金的Fe能增加Pd的电子云密度,使催化剂中Pd0的含量增加,因此,提高了Pd催化剂对氧还原的电催化性能。
[1]Appleby A J,Lloyd A C,Dyer C K.Sci.Am.,1999,281(1): 72-77
[2]Zhang L,Zhang J J,Wilkinson D P,et al.J.Power Sources, 2006,156(2):171-182
[3]Gasteiger H A,Kocha S S,Sompalli B,et al.Appl.Catal.B: Environ.,2005,56(1-2):9-35
[4]Demirci U B.J.Power Sources,2007,173(1):11-18
[5]Wang B.J.Power Sources,2005,152(1):1-15
[6]LI Xu-Guang(李旭光),XING Wei(邢巍),LU Tian-Hong(陆天虹),et al.Chem.J.Chinese Universities(高等学校化学学报),2003,7(24):1246-1250
[7]Shao M H,Sasaki K,Adzic R R.J.Am.Chem.Soc.,2006, 128(11):3526-3527
[8]Song S Q,Wang Y,Tsiakaras P,et al.Appl.Catal.B:Environ., 2008,78(3/4):381-387
[9]Jin Y X,Ma C N,Shi M Q,et al.Int.J.Electrochem.Sci., 2012,7(4):3399-3408
[10]Wang H,Ji S,Wang W,et al.Int.J.Electrochem.Sci., 2012,7(4):3390-3398
[11]Trinh Q T,Yang J H,Lee J Y,et al.J.Catal.,2012,291:26-35
[12]Pires F I,Villullas H M.Int.J.Hydrogen Energy,2012,37 (22):17052-17059
[13]Li A Z,Zhao X,Hou Y N,et al.Appl.Catal.B:Environ., 2012,111:628-635
[14]Zhang Z Y,More K L,Sun K,et al.Chem.Mater.,2011,23 (6):1570-1577
[15]Yin S B,Cai M,Wang C X,et al.Energy Environ.Sci., 2011,4(2):558-563
[16]Neergat M,Gunasekar V,Rahul R.J.Electroanal.Chem., 2011,658(1/2):25-32
[17]Alexeyeva N,Sarapuu A,Tammeveski K,et al.Electrochim. Acta,2011,56(19):6702-6708
[18]Yang J H,Zhou W J,Cheng C H,et al.Appl.Mat.Interfaces, 2010,2(1):119-126
[19]Wang W,Wang R F,Ji S,et al.J.Power Sources,2010,195 (11):3498-3503
[20]Tang Y W,Cao S,Chen Y,et al.Appl.Surf.Sci.,2010,256 (13):4196-4200
[21]Yeh Y C,Chen H M,Liu R S,et al.Chem.Mater.,2009,21 (17):4030-4036
[22]Tarasevich M R,Zhutaeva G V,Bogdanovskaya V A,et al. Electrochim.Acta,2007,52(15):5108-5118
[23]Xu J,Lü X S,Li J D,et al.J.Hazard.Mater.,2012,225:36-45
[24]Pan Y,Zhang F,Wu K,et al.Int.J.Hydrogen Energy, 2012,37(4):2993-3000
[25]Wang C,Markovic N M,Stamenkovic V R.ACS Catal., 2012,2(5):891-898
[26]Vondrova M,Burgess C M,Bocarsly A B.Chem.Mater., 2007,19(9):2203-2212
[27]Wang R,Liao S,Fu Z,et al.Electrochem.Commun.,2008, 10(4):523-526
[28]Li W Z,Haldar P.Electrochem.Commun.,2009,11(6):1195-1198
[29]Radmilovic V,Gasteiger H A,Ross P N.J.Catal.,1995, 154(1):98-106
[30]Antolini E,Cardellini F.J.Alloys Compd.,2001,315(1/2): 118-122
[31]Zhang L,Lee K,Zhang J.Electrochim.Acta,2007,52(9): 3088-3094
[32]Wang W,Zheng D,Du C,et al.J.Power Sources,2007,167 (2):243-249
[33]Tang Y,Zhang L,Wang Y,et al.J.Power Sources,2006, 162(1):124-131
[34]Tominaka S,Mommab T,Osaka T.Electrochim.Acta,2008, 53(14):4679-4686
[35]Dumbuya K,Denecke R,Steinruck H P.Appl.Catal.A: Gen.,2008,348(2):209-213
[36]Zhang L,Tang Y,Bao J,et al.J.Power Sources,2006,162 (1):177-179
[37]Persson K,Ersson A,Jansson K,et al.J.Catal.,2005,231 (1):139-150
Carbon Supported Alloy Pd-Fe Catalyst:Preperation and Electrocatalytic Activity for Oxygen Reduction
WANG Yan-En1CAO Shuang*,1LIU Shu-Jing1FENG Tao1LIU Ning1TANG Ya-Wen*,2LU Tian-Hong2
(1Department of Morden Science&Technology,College of Science,Agricultural University of Hebei,Baoding,Hebei 071000,China)
(2School of Chemistry and Materials Science,Nanjing Normal University,Nanjing 210097,China)
The Pd-Fe/C catalyst was prepared by the complexing reduction method using NH4Cl as the complex agent at the low temperature.The high alloy Pd-Fe/C catalyst Pd and Fe could be prepared at low temperature due to the complex formation by NH4Cl and Pd,which leads to a negative shift for the reduction potential of PdCl2,making the reduction potential of PdCl2closer to that of FeCl3.The XPS results show that the alloying of Pd with Fe could affect the binding energies of Pd and increase the content of Pd0in the catalyst.Thus,the electrocatalytic activity of the Pd-Fe/C catalyst obtained for the oxygen reduction is higher than that of the Pd/C catalyst prepared with the same method.Furthermore,this Pd-Fe/C catalyst has no electrocatalytic activity for the methanol oxidation.
catalyst;complexing reduction method;Pd-Fe/C catalyst;reduction potential;high alloy;oxygen reduction
O613.71
A
1001-4861(2015)01-0023-06
10.11862/CJIC.2015.024
2014-05-12。收修改稿日期:2014-10-17。
国家自然科学基金项目(No.21073094,21273116,61171015)和江苏高校优势学科建设工程(No.10KJB150007)资助项目。*
。E-mail:caoshuang96@163.com
猜你喜欢
云南化工(2020年11期)2021-01-14
科学(2020年4期)2020-11-26
矿产综合利用(2020年1期)2020-07-24
科学(2020年4期)2020-01-11
数学物理学报(2019年5期)2019-11-29
中国有色金属学报(2018年2期)2018-03-26
环境保护与循环经济(2017年1期)2017-09-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
潍坊学院学报(2016年6期)2016-04-18
中国资源综合利用(2016年7期)2016-02-03
