
基于全自动毛细管电泳技术建立的单核细胞增生李斯特氏菌MLVA分型方法
2015-05-09 01:03李秀娟崔玲玲高伟利
中国人兽共患病学报 2015年9期
李秀娟,崔玲玲,赵 冬,潘 琢,高伟利
基于全自动毛细管电泳技术建立的单核细胞增生李斯特氏菌MLVA分型方法
李秀娟1,崔玲玲2,赵 冬1,潘 琢1,高伟利1
目的 建立针对食品来源的单核细胞增生李斯特氏菌(Listeria monocytogenes,Lm)分离株的多位点串联重复序列分型(Multiple-Locus Variable number tandem repeat Analysis ,MLVA)方法,为暴发确认和溯源检测提供实验室支持。方法 对2005—2014年间分离自食品的91株Lm进行14个可变数目串联重复序列(Variable Number of Tandem Repeats, VNTR)位点的检测,评估最优检测位点组合并分析检测结果。结果 通过采用软件分析,由LMV1、LMV2、LMV7、Lm10、Lm11、Lm23、LM-TR6、TR3和Lm15 等9个VNTR位点组成的位点组合为最优MLVA检测位点,可以将91株Lm分离株分为70个型别,分型能力达到0.987 1。结论 本研究建立的基于全自动毛细管电泳的由9个检测位点组成的Lm的MLVA分型方法,具有操作简便、快速、结果客观、操作标准化、易于在不同实验室间比较的优势,可作为一线检测方法用于李斯特菌病的暴发确认和溯源检测。
单核细胞增生李斯特氏菌;多位点串联重复序列分型;毛细管电泳
李斯特菌属为革兰染色阳性兼性厌氧无荚膜杆菌,有7个菌种,分别为单核细胞增生李斯特氏菌(L.monocytogenes,Lm)、绵羊李斯特菌(Listeriaiuanuii)、英诺克李斯特菌(Listeriainnocua)、威尔斯李斯特菌(Listeriainnocua)、西尔李斯特菌(Listeriaseeligeri)、格氏李斯特菌(Listeriagrayi)和默氏李斯特菌(Listeriamurrayi)[1-2]。Lm是唯一能引起人类疾病的李斯特菌,作为一种人畜共患病的病原菌,它能引起人、畜的李氏特菌病,感染后主要表现为败血症、脑膜炎和单核细胞增多[3]。尽管李斯特菌病的发病率相对较低,但对老年人和有免疫缺陷的感染者,致死率可达30%[4]。李斯特菌病主要通过食源性途径感染,可引起散发和暴发事件的发生。由于该病的发病率较低,潜伏期长,最长可达71 d,导致在确认李斯特菌病暴发以及追查污染源方面有较大的困难。而建立可靠的Lm分型方法,拥有分型数据库对于暴发确认和溯源检测具有十分重要的意义。本研究旨在通过对分离自食品的Lm的14个可变数目串联重复序列(Variable Number of Tandem Repeats, VNTR)位点的检测,通过评估检测位点组合,建立针对食品中Lm分离株的MLVA分型方法,确定优势型别,建立MLVA分型数据库,为以后的溯源检测和暴发确认提供实验室资料支持。
1 材料和方法
1.1 材料
1.1.1 菌株来源 91株Lm为2005—2014 年分离自生禽肉、生畜肉、水食产品、速冻米面制品及凉拌色拉,均经李斯特菌API Listeria生化鉴定试剂条检测确认。其中43株Lm分离株由石家庄市疾病预防控制中心分离,48株由河北省疾病预防控制中心馈赠。
1.1.2 主要设备与试剂 9700型PCR仪(ABI公司),Qiaxcel型全自动毛细管电泳仪(Qiagen 公司),GoTaq®Colorless Master Mix(Promega公司),高分辨率的毛细管电泳卡夹(DNA High Resolution Kit Gel Cartridge,Qiagen公司)。
1.2 方法
1.2.1 DNA模板制备 从血平板上挑取过夜培养的Lm新鲜菌落至400 μL×TE(pH8.0)缓冲液中,震荡混匀,煮沸5 min,冰上冷却,10 000 r/min离心5 min,取上清于-20 ℃保存备用。
1.2.2 MLVA检测 本研究所用MLVA检测位点及引物参考文献[5-9]。为了避免检测位点的重复,全部22个位点均经过比对确认,最后确认14个位点作为筛选位点,见表1。所有14个位点均采用单重PCR检测,PCR体系:2 ×Gotaq®Colorless Master Mix 12.5 μL,上、下游引物(10 μmol/L)各1 μL,DNA模板1 μL,加水补足体积至25 μL。PCR参数:95 ℃ 5 min;94 ℃ 30 s,50 ℃ 30 s,72 ℃ 40 s,35个循环;72 ℃ 5 min。采用高分辨率的毛细管电泳卡夹经全自动毛细管电泳仪检测PCR扩增结果。
1.2.3 串联重复序列数目的确认 对每个位点的不同长度的扩增产物均送上海生工公司进行双向测序,参考串联重复序列分析软件Tandem Repeat Finder,确定重复序列数目。对于无扩增产物的位点,重复序列数目确定为0;有扩增产物而无重复序列的位点,相应重复序列数目为1;其余所有菌株重复序列数目均为实际重复序列数目+1。
1.2.4 分子血清型检测 参考文献[10],采用一个多重PCR进行Lm的分子血清学检测。每次PCR均使用阳性对照,阳性对照菌株为09M692(1/2c)和11M5102(4b),由澳大利亚昆士兰州法医与科学研究所馈赠。
1.2.5 谱系检测 参考文献[11],基于毒力基因actA序列第259位至437位氨基酸的测定,采用Mega5.0软件将各序列与文献中公布的谱系特异性氨基酸序列进行比较分析。
1.3 数据分析 采用辛普森指数(Simposon’s index of diversity,DI)作为计算分辨力的方法[12],检测位点的最优组合计算采用Optimal Combination Finder (OCT)软件进行[13]。采用BioNumerics Version 6.6软件对MLVA分型结果以UPGMA方法构建进化树。
2 结 果
2.1 14个VNTR位点检测结果 通过对14个VNTR位点的PCR扩增,绝大多数位点均可扩增出唯一的目的条带,但也有些位点无扩增产物(表1)。在91 株Lm中,LM-TR6位点有70株菌无扩增产物,其次为Lm11、LMV9、LMV7和Lm23位点,菌株数分别为17、14、1和1株。Lm11位点无扩增产物的17株菌株均为1/2a血清型,LMV9位点无扩增产物的14株菌株均为1/2b血清型,LMV7和Lm23位点无扩增产物的菌株分别为1 株1/2a血清型和1 株1/2b血清型;而LM-TR6位点在28株1/2b血清型菌株中均无扩增产物,在15株4b型菌株中有14株无扩增产物,在1/2a型和1/2c型中,分别有27株和1株菌株无扩增条带。其余9个位点均有不同长度的扩增产物。在14个检测位点中LMV2位点的分型能力最强,将91株菌分为15个型别,分型指数为0.891 5,其次为Lm23,分型指数为0.879 1。而LM-TR2位点的分型能力最弱,91株菌为同一型别。
2.2 MLVA位点最优组合的选择 通过采用OCT软件计算由2-10个检测位点组成的所有组合的分型能力(表2),发现随着组合位点数目的增多,大体呈现分型能力提高的趋势,但在组合位点达到9个和10个时,分型能力并无增加,而是停留在0.987 1。由此可见,由LMV1、LMV2、LMV7、Lm10、Lm11、Lm23、LM-TR6、TR3和Lm15等9 个位点组成的MLVA检测体系是最优的检测位点组合,将91株菌分为70个型别,分型能力达到0.987 1。
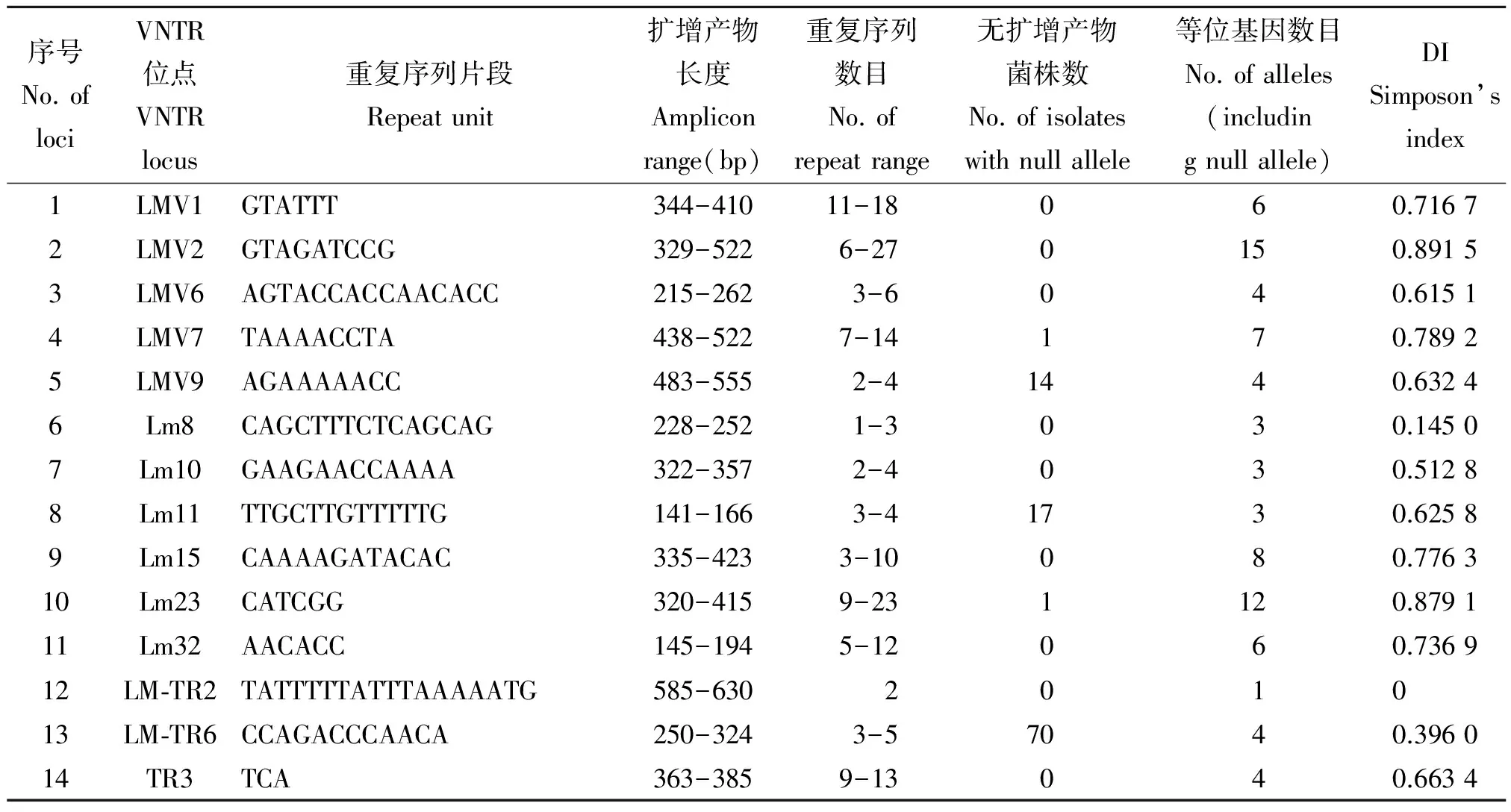
表1 14个VNTR位点检测结果
2.3 最优的9个VNTR位点组合对不同血清型菌株的分型检测 利用分子血清分型方法,可将91株Lm分为四种血清类型,分别为1/2a型(1/2a和3a)41株、1/2b型(1/2b、3b和7)28株、1/2c型(1/2c和3c)7株 和4b型(4b、4d和4e)15株。新建立的最优的9个位点组合对不同血清型Lm菌株的分型能力均较好,DI值除1/2a型为0.948 7以外,余均大于0.95(表3)。各血清型的主要基因型别非常集中,且该主要基因型同时是该血清型的独特基因型,在其他血清型中均未检出。
2.4 进化树分析 采用BioNumerics Version 6.6软件以UPGMA方法构建进化树(图1),进化树分析可将91株Lm分为两大分支,分别为谱系Ⅰ和谱系Ⅱ,谱系Ⅰ由1/2b和4b血清型的菌株组成,谱系Ⅱ由1/2a和1/2c血清型的菌株组成。谱系Ⅰ的1/2b和4b血清型混杂在一起,不能进一步区分。而谱系Ⅱ中的7株1/2c血清型菌株单独聚集成一支。
3 讨 论

图1 以UPGMA方法对MLVA检测结果构建进化树
Fig.1 UPGMA dendrogram of MLVA results for 91L.monocytogenesisolates
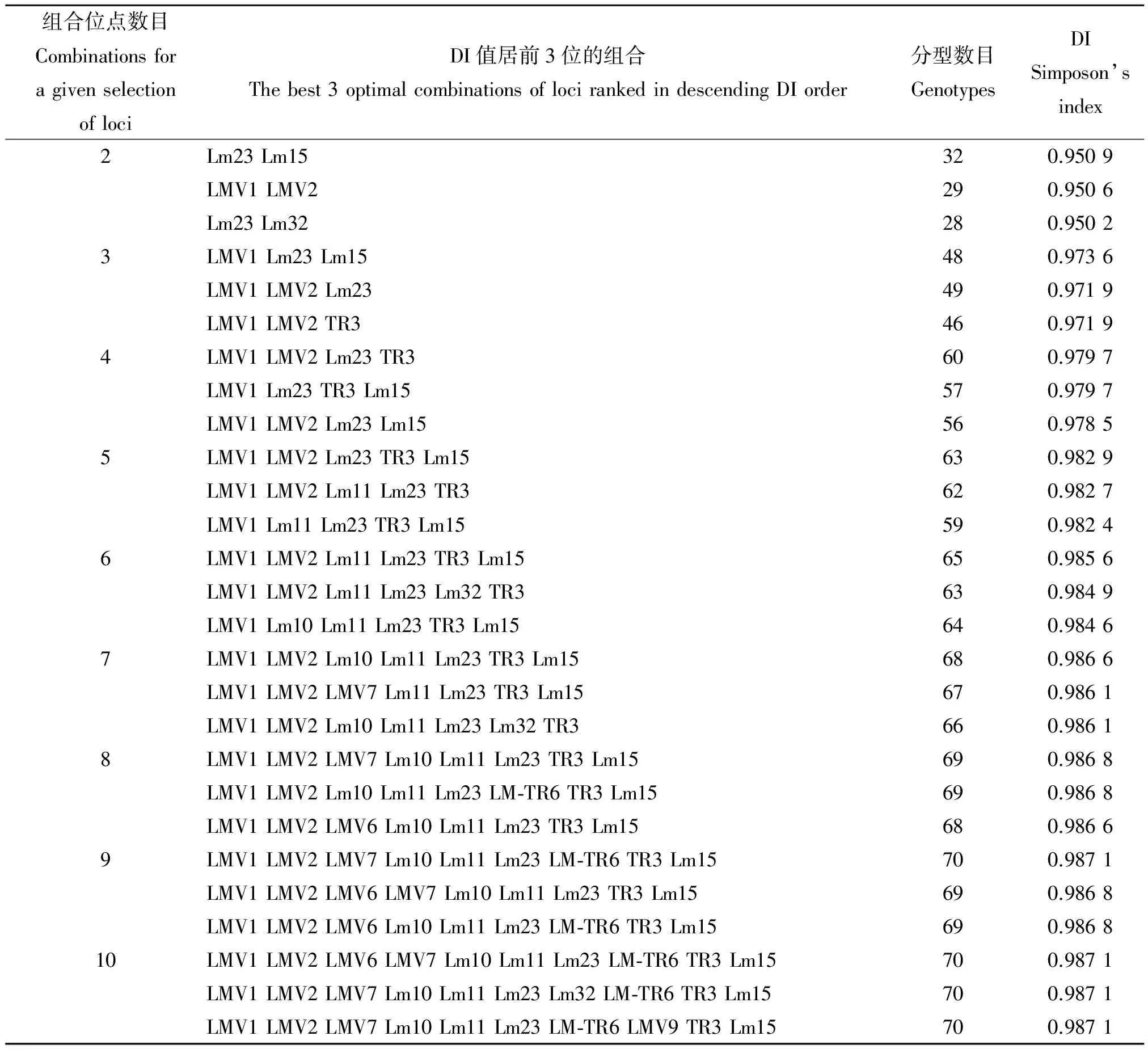
表2 不同位点组合的分型能力
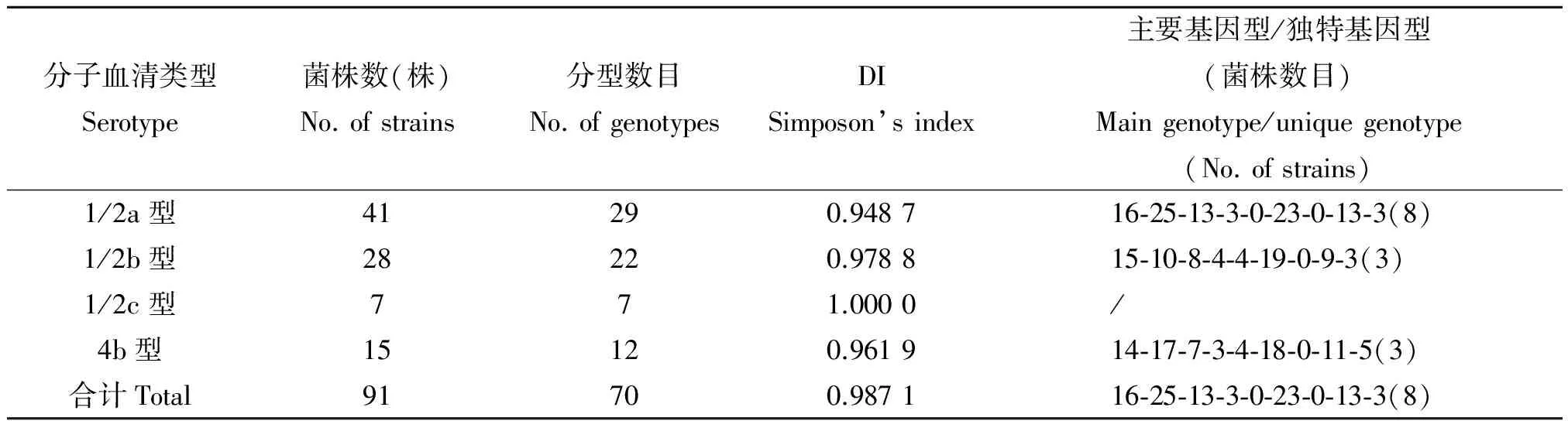
表3 新建立MLVA方法对不同血清型菌株的分型结果
细菌分型检测的主要意义在于暴发确认和污染源追踪,根本目的是为了确认关联菌株。PFGE分型方法采用单一酶切,并不能很好的分辨不同的Lm菌株,需同时进行两种酶切才能增加分辨力。PFGE虽目前已作为金标准用于分型和暴发溯源检测,但该方法的缺点如仪器昂贵、方法难于标准化、结果分析不可避免的主观化、结果分析需采用专门软件等,已明显限制了该方法的推广应用。目前已有多篇文献报道,MLVA分型方法可区分流行病学相关菌株和非相关菌株[7,14-16]以及相关克隆和非相关克隆[17]。2007年,Murphy[8]等首次报道了采用MLVA方法进行Lm的分型检测,至目前也已有多篇文献对该方法的建立进行了报道。但由于各文献采用不用的检测位点和组合,且没有稳定的标准对位点组合进行评估,因此到目前为止还没有一套稳定的位点组合作为Lm的MLVA分型检测标准。在目前报道的一些检测位点中,有些位点的分型能力较弱,有些检测位点则互相重叠,另还有一些位点出现较多的无扩增现象。筛选合适的检测位点组合,建立标准化的位点组合和分型策略,对分型检测结果的确定和比对具有十分重要的意义。
Chenal-Francisque等[17]曾根据VNTR位点在染色体中位置的不同,选用了18个VNTR位点作为检测对象,根据每个位点扩增结果是否满意,即是否有非特异性扩增,是否有无扩增产物的菌株出现,串联重复序列数目是否容易计算等原因,选择了11个VNTR位点作为优化位点组合,但由于未考虑位点组合不同会导致不同分型结果,因此该方法的DI值只有0.945,低于PFGE方法。笔者曾在澳大利亚采用该14个位点对澳大利亚的183株Lm菌株进行MLVA检测,DI值最高可达0.991 4,最优组合同样也是9个位点的组合。其中与本研究相同的位点有LMV1、LMV2、LMV7、Lm10 Lm11、Lm23、LM-TR6,不同的位点是以TR3、Lm15替换了LMV6和Lm32。提示菌株来源的不同可能会导致不同的检测结果,即需要不同的位点组合进行MLVA分型的检测。Lindstedt[6]也曾报道采用相同的位点组合针对挪威和瑞典两个国家的Lm分型时显示出不同的分辨能力,作者认为分辨能力的不同是由于选取菌株来源不同所致。由于MLVA检测方法的建立是基于具有较高变异率的VNTR位点,因此可能会表现出由于菌株来源不同,而导致分型能力的差异,同时也会出现不同的位点组合导致不同的分型能力的结果,因此建立一套适合我国的、稳定的Lm的MLVA方法在李斯特菌病的防控上具有十分重要的意义。本研究最初采用14个VNTR位点对91株Lm进行了检测,通过采用OCT软件进行不同位点组合的DI值计算,考虑了不同位点组合对分辨力的影响,确定了由9个检测位点组成的Lm 的MLVA检测体系。
多篇文献报道[7-10,14]显示LMV2位点的分型能力最强,这与本研究的结果相同,提示该位点变异较大。Sperry报道[9]LM-TR6位点不能扩增1/2b、3b和4b型菌株,本研究结果同样显示该位点在1/2b血清型中不存在,而分子血清型为4b的15株菌株中,同样有14株无扩增产物,而仅有的一株有扩增产物的菌株有可能为4d或4e型菌株,还有待进一步做血清凝集证实。而lm11位点在Sperry[9]首次使用该位点进行MLVA分析时,并未显示出所有的1/2a菌株均无扩增条带,本研究显示的17株1/2a菌株均无扩增条带,可能为样本量少,也可能为菌株来源差异所致。LMV9位点在14株1/2b菌株中无扩增条带,这与Lindstedt[6]文献中显示的部分1/2b菌株无扩增条带的结果相同,但目前由于文献中菌株量较少,尚无能确定该位点仅在1/2b菌株中会出现缺失。
在17种Lm的血清型中,引起人类李斯特菌病的主要为1/2a、1/2b和4b型菌株。Lindstedt[6]报道采用针对4b型菌株的VNTR位点进行的MLVA分析,检测结果经进化树分析可见4b型菌株形成不同于1/2血清型的单一簇,可以用于快速区分4b型菌株。本研究采用优化的9个位点进行MLVA分型检测,对不同血清型均有很好的分辨力,但进化树分析并不能进一步将4b型菌株区分开来,但能区分谱系Ⅰ和谱系Ⅱ的菌株,提示该结果同样也可为暴发和菌株的确认提供支持。
本研究优化的9个位点,虽然有LM-TR6等可产生较多无扩增条带的位点,但在确定整体优化组合时,LM-TR6位点的加入可使分型数目由69增加至70,增加了一个基因型,DI值达最大。本研究结果还显示,Lm23与Lm15两个位点的组合,DI值即可达到0.950 9,可满足区分不同菌株的能力,提示当检测菌株数量较多,时间较紧时,可以通过减少检测位点的数量,同样可达到很好的分析结果,可作为实验室的一线方法用于李斯特菌病暴发调查和监测。
本研究所用的Lm均来源于食品,虽然来源较单一,但采用本研究确定的MLVA方法仍可将91株菌分为70个基因型别,分型能力达0.987 1,可用于不同血清型Lm菌株的分型检测。该方法建立在普通PCR基础之上,联合使用高分辨率的全自动毛细管电泳,具有操作简便、快速、价廉、高通量、重复性好,可对食品样品直接进行检测[16]的优点,另外该方法将检测结果数字化,而非采用图片分析形式,减少了在结果分析过程中的主观影响,更易实现结果客观化和操作标准化,因此便于不同实验室间的结果比较。据此可建立类似于PulseNet网络的在线数据库,进行全国范围的结果比较,从而为该菌在大范围内的暴发事件确认提供支持。
[1]Jeffers GT, Bruce JL, McDonough PL, et al. Comparative genetic characterization ofListeriamonocytogenesisolates from human and animal listeriosis cases[J]. Microbiol, 2001, 147 (5): 1095-1104.
[2]Snehal J, Mrinal B, Enzo A. Methods used for the detection and subtyping ofListeriamonocytogenes[J]. J Microbiol Methods, 2012, 88 (3): 327-341. DOI: 10.1016/j.mimet.2012.01.002
[3]Xie MH, Yang XM, Lv XB. Investigation on pollution status oflisteriamonocytogenesin food[J]. Hubei Prev Med, 1999, 10 (2): 31. (in Chinese) 谢茂慧, 杨晓敏, 吕斌. 食品中单核细胞增生李斯特菌污染的调查研究[J]. 湖北预防医学杂志, 1999, 10 (2): 31.
[4]Mead PS, Slutsker L, Dietz V. Food-related illness and death in the United States[J]. Emerg Infect Dis, 1999, 5(5): 607-625. DOI: 10.3201/eid0506.990624
[5]Li X, Huang B, Eglezos S, et al. Identification of an optimized panel of variable number tandem-repeat (VNTR) loci forListeriamonocytogenestyping[J]. Diagn Microbiol Infect Dis, 2013, 75 (2): 203-206. DOI: 10.1016/j.diagmicrobio.2012.11.007
[6]Lindstedt BA, Tham W, Danielsson-Tham ML, et al. Multiple-locus variable-number tandem-repeats analysis ofListeriamonocytogenesusing multicolour capillary electrophoresis and comparison with pulsed-field gel electrophoresis typing[J]. J Microbiol Methods, 2007, 72 (2): 141-148.DOI: 10.1016/j.mimet.2007.11.012
[7]Miya S, Kimura B, Sato M, et al. Development of a multilocus variable-number of tandem repeat typing method forListeriamonocytogenesserotype 4b strains[J]. Int J Food Microbiol, 2008, 124 (3): 239-249. DOI: 10.1016/j.ijfoodmicro.2008.03.023
[8]Murphy M, Corcoran D, Buckley JF, et al. Development and application of multiple-locus variable number of tandem repeat analysis (MLVA) to subtype a collection ofListeriamonocytogenes[J]. Int J Food Microbiol, 2007, 115 (2): 187-194. DOI: 10.1016/j.ijfoodmicro.2006.10.022
[9]Sperry KE, Kathariou S, Edwards JS, et al. Multiple-locus variable-number tandem-repeat analysis as a tool for subtypingListeriamonocytogenesstrains[J]. J Clin Microbiol, 2008, 46(4): 1435-1450. DOI: 10.1128/JCM.02207-07
[10]Doumith M, Buchrieser C, Glaser P, et al. Differentiation of the majorListeriamonocytogenesserovars by multiplex PCR[J]. J Clin Microbiol, 2004, 42 (8): 3819-3822. DOI: 10.1128/JCM.42.8.3819-3822.2004
[11]Wiedmann M, Bruce JL, Keating C, et al. Ribotypes and virulence gene polymorphisms suggest three distinctListeriamonocytogeneslineages with differences in pathogenic potential[J]. Infect Immun, 1997, 65(7): 2707-2716.
[12]Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity[J]. J Clin Microbiol, 1988, 26 (11): 2465-2466.
[13]Wang X, Huang B, Blair B, et al. Selection of optimal combinations of loci by the Optimal Combination Finder computer program from a group of variable number tandem repeat loci for use inStaphylococcusaureusfood poisoning case investigations[J]. J Med Microbiol, 2012, 61 (5): 631-639.DOI: 10.1099/jmm.0.040287-0
[14]Lunestad BT, Truong TT, Lindstedt BA. A multiple-locus variable-number tandem repeat analysis (MLVA) ofListeriamonocytogenesisolated from Norwegian salmon-processing factories and from listeriosis patients[J]. Epidemiol Infect, 2013, 141: 2101-2110. DOI: 10.1017/S0950268812002750
[15]Saleh-Lakha S, Allen VG, Li J, et al. Subtyping of a large collection of historicalListeriamonocytogenesstrains from Ontario, Canada, by an improved multilocus variable-number tandem-repeat analysis (MLVA)[J]. Appl Environ Microbiol, 2013, 79 (20): 6472-6480. DOI: 10.1128/AEM.00759-13
[16]Chen S, Li J, Saleh-Lakha S, et al. Multiple-locus variable number of tandem repeat analysis (MLVA) ofListeriamonocytogenesdirectly in food samples[J]. Int J Food Microbiol, 2011, 148 (1): 8-14. DOI: 10.1016/j.ijfoodmicro.2011.04.014
[17]Chenal-Francisque V, Diancourt L, Cantinelli T, et al. Optimized multilocus variable-number tandem-repeat analysis assay and its complementarity with pulsed-field gel electrophoresis and multilocus sequence typing forListeriamonocytogenesclone identification and surveillance[J]. J Clin Microbiol, 2013, 51(6): 1868-1880. DOI: 10.1128/JCM.00606-13
Development of a multiple-locus variable number of tandem repeat typing method forListeriamonocytogenesupon the capillary electrophoresis
LI Xiu-juan1,CUI Ling-ling2,ZHAO Dong1,PAN Zhuo1,GAO Wei-li1
(1.DepartmentofMicrobiology,ShijiazhuangCenterforDiseaseControlandPrevention,Shijiazhuang050011,China; 2.HebeiNormalUniversity,Shijiazhuang050000,China)
In order to develop a multiple-locus variable number tandem repeat analysis (MLVA) method forListeriamonocytogenes(L.monocytogenes), a total of 14 variable number of tandem repeats (VNTR) loci were used to detect the 91L.monocytogenesisolated from food. Results showed that the optimal combination of loci consisted of 9 loci (LMV1, LMV2, LMV7, Lm10, Lm11, Lm23, LM-TR6, TR3, Lm15), which produced the high level of discriminatory ability with Simpson index of 0.987 1 for foodborneL.monocytogenes, could divided 91L.monocytogenesisolates into 70 subtypes. This method required only one conventional PCR followed by automated capillary electrophoresis, providing high discriminatory ability. It was simple, easy to perform, relatively fast, inexpensive, objective, standardized operating and could provide conveniently interlaboratory comparisons, which would be useful in outbreak investigations and listeriosis surveillance as a first line screening method.
Listeriamonocytogenes; multiple-locus variable number tandem repeat analysis (MLVA); capillary electrophoresis
10.3969/j.issn.1002-2694.2015.09.012
1.石家庄市疾病预防控制中心,石家庄 050011 2.河北师范大学,石家庄 050000; Email:wsws1120@126.com
R378.99
A
1002-2694(2015)09-0839-06
2015-03-12;
2015-06-12
猜你喜欢
传染病信息(2022年6期)2023-01-12
首都食品与医药(2022年21期)2022-12-06
艺术品鉴(2022年16期)2022-07-09
医药前沿(2021年25期)2021-10-16
河池学院学报(2021年1期)2021-07-10
景德镇陶瓷(2021年1期)2021-03-24
养猪(2020年6期)2021-01-27
当代陕西(2019年19期)2019-11-23
小小说月刊(2013年6期)2013-05-14
琴童(2009年2期)2009-02-26
