
地表水中敌百虫测定技术难点探讨
2015-04-26 00:55:48许秀艳滕恩江吕怡兵
中国环境监测 2015年2期
陈 烨,许秀艳,王 超,刀 谞,滕恩江,吕怡兵
中国环境监测总站,国家环境保护环境监测质量控制重点实验室,北京 100012
目前,农业生产中使用的农药主要分为4大类:有机氯农药、有机磷农药、氨基甲酸酯类农药和拟除虫菊酯类农药。其中,有机磷农药的使用比例占所有农药的70%以上,在农业生产中的广泛应用,也使其成为重要的环境污染物。
与其他农药类别相比,有机磷农药的性质比较特殊,很多组分的化学性质不稳定,对光、热、氧、碱等环境条件都较为敏感,且极性差别很大[1]。因此,有机磷农药的分析难度也远大于其他农药。
在有机磷农药组分中,同时具有不稳定性和强极性的敌百虫,在分析测定的各阶段都面临一系列特殊问题。样品采集和保存阶段,不稳定性使得敌百虫很容易发生分解;前处理阶段,强极性使得从水体中富集敌百虫的难度加大,也可能存在目标物分解的情况;仪器分析阶段,不稳定性限制了可直接用于敌百虫分析的仪器种类。
鉴于上述原因,实际分析中常会出现敌百虫测定结果不理想的情况(如回收率差、灵敏度低等),甚至出现目标物丢失、定性定量错误等问题[2]。因此,充分把握敌百虫的特性,全面认识可能存在的问题,对准确测定敌百虫具有十分重要的意义。研究从敌百虫的基本性质和降解途径入手,对地表水中敌百虫残留分析各阶段中可能出现的问题及解决方法进行了综述。
1 敌百虫的基本性质
1.1 理化性质[3]
敌百虫(C4H8Cl3O4P)属于有机磷杀虫剂,是一种带有微弱特殊气味的无色晶体。其基本理化性质为熔点78.5℃;20℃下蒸汽压为0.21 MPa,25℃下为0.5 MPa;20℃下,辛醇-水的分配系数为 0.43,亨利系数为 4.4 × 10-7Pa·m3/mol,比重为1.73,水中溶解度为120 g/L;易溶于常见的有机溶剂。
敌百虫稳定性较差,易发生水解和脱氯化氢反应,尤其在受热和pH>6的条件下,降解反应进行得更快。22℃下降解半衰期为510 d(pH=4),46 h(pH=7)和小于30 min(pH=9)。碱性条件下,敌百虫快速水解为敌敌畏;酸性条件下,敌百虫缓慢水解发生甲酯基团的裂解反应。
1.2 降解途径[4]
图1显示了敌百虫在环境中的降解途径。
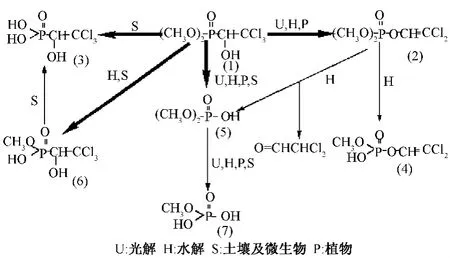
图1 敌百虫在环境中的降解途径
由图1可见,敌百虫在环境中存在多种降解途径,光解、水解、土壤、植物及微生物作用都可使其发生分解。环境水体中,敌百虫的降解途径主要为水解和光解。当pH≥5.5时,敌百虫的水解速度非常快,其脱去氯化氢,经分子重排生成敌敌畏。在紫外线的照射下,敌百虫很快分解为敌敌畏和其他2种降解产物,后者在持续的照射下还将继续分解。
2 样品采集和保存
样品采集和保存条件将直接影响分析结果的可靠性。由于敌百虫很不稳定,对热、碱等环境条件都较为敏感,故采集地表水样时除了满足一般性的采集和保存要求外,还有其他需要注意的方面。
国内外标准中,明确涉及地表水中敌百虫测定的样品采集和保存条件并不多(表1),其中的规定也不尽相同,甚至还存在相互矛盾之处。主要表现在采样容器是否需用水样预先荡洗、样品保存时间和pH方面。现有文献资料中,有关这方面的研究也基本处于空白状态。

表1 现行标准中有关敌百虫测定的水样采集和保存规定[5-9]
采样瓶是否需用水样预先荡洗,主要是考虑容器内壁对目标物是否有吸附作用,这与容器的清洗方式、内壁的表面性质及样品中目标物的种类浓度都直接相关[10]。由表1可见,国内现行标准中,关于这方面的规定尚不统一。但EPA和ISO的标准中,对于采样瓶是否需用水样预先荡洗的规定是明确且一致的:不要用水样荡洗玻璃采样瓶,以免目标物吸附于瓶壁。
关于保存时间,理想的情况是采样后立即进行分析,尤其是不稳定的待测物。如不能尽快分析,可将样品先行进行前处理,前处理后得到的提取液比原样品的有效保存时间更长。
另外,从降解半衰期数据可知,对敌百虫来说,保存pH直接影响保存时间的长短。为尽量延缓敌百虫的降解,应使样品在pH<5.5的酸性条件下保存。
当然,由于样品类型具有多样性,同一采集和保存方法不可能同时满足各类水体中敌百虫残留测定的要求。建议在实际工作中,采样前应根据样品性质、环境条件以及待测目标物等具体条件,对采集和保存方法进行验证,然后酌情选用[11-12]。
一般来说,从开始进行样品采集和保存条件实验到确定适宜的采集和保存方法,需要经过如下步骤[13]:①对检出限、精密度、准确度等方法性能指标进行测试,确保分析方法可以很好地反映测定对象的实际情况。②测定样品中目标物的含量即定值,并通过精密度指标衡量样品的均匀性,保证后续实验数据的可比性。③将样品分组,按照设定的采集和保存方案进行实验。
3 常用的前处理方法
3.1 液-液萃取法
液-液萃取法是经典的前处理方法,在实际环境监测工作中应用的时间最长、范围最广。由于敌百虫的极性大、水溶性强,直接用有机溶剂萃取的提取率极低,故液-液萃取时,需要采用间接测定的方法[5]。将敌百虫转化为敌敌畏,通过测定敌敌畏的含量换算得到敌百虫的含量。由于敌敌畏在碱性条件下也能继续水解,因此在转化反应中应严格控制温度、时间和pH等反应条件[14]。具体参考条件为[5]调节水样pH至9.6,倒入锥形瓶,盖好瓶塞,于50℃水浴进行碱解反应,反应过程中不断摇动锥形瓶。15 min后取出锥形瓶,冷却至室温后,调节pH至6.5,进行液-液萃取。
实际水样中往往还含有除敌百虫以外的其他有机磷农药组分,在提取敌百虫前,需要先将其他组分在中性条件下萃取出来,然后再进行敌百虫的转化反应。由于敌百虫仅在酸性条件下较为稳定[4],在萃取其他组分的过程中,很容易发生敌百虫的部分分解,造成其测定值偏低。另外,如果水样中本身就存在敌敌畏,则敌百虫的提前部分分解,还会造成敌敌畏的测量值偏离实际。
3.2 固相萃取法
固相萃取法通过吸附剂选择性保留样品中的目标物,实现对样品的分离、纯化和富集。
吸附剂的选择直接影响目标物的富集效果,应综合考虑目标物性质、样品基质和干扰物的情况,选择对目标物有较强保留能力的吸附剂。对敌百虫的富集来说,还要充分考虑其强极性特性,选择可对极性物质提供优异反相保留容量的固相萃取吸附剂。
最常见的C18固相萃取吸附剂,已被用于地表水样品中敌百虫等有机磷农药的富集。加标量在1~5 μg/L水平时,C18固相萃取盘用于富集水样中敌百虫等10种有机磷农药的加标回收率为50% ~96%[15]。萃取河水中的敌百虫时,检出限为0.01 μg/mL,回收率为87.6% ~101.3%,相对标准偏差为1.70% ~2.77%[16]。
冯复来等[17]比较了 Waters SEP-PAK C18、IST C18和Whatman ODS-5 3种固相萃取柱的富集效果。其中,Waters SEP-PAK C18对敌百虫的富集回收率达70%以上,其他2种萃取柱则都在10%以下。
Shmamura等[18]研究了 Sep-pak PS-2 固相萃取柱对水样中包括敌百虫在内的共22种农药的富集情况。对纯水和河水2种水样进行加标回收实验,两者的回收率分别为 81% ~103%和71% ~127%。
目前,常见的固相萃取填料类型共有4类:键合硅胶,高分子聚合物,吸附性填料,混合型及专用柱。现有文献中使用较多的还是常见的硅胶基质吸附材料,这类填料种类多,可选择的范围广,但存在对极性化合物保留不足等问题。高分子聚合物填料中引入了极性官能团,具有比硅胶基质更强的吸附性,对各类极性、非极性化合物均具有较好的保留作用,应用这类吸附材料富集样品中的敌百虫将是未来的研究发展方向。
3.3 固相微萃取法
固相微萃取法是集采样、萃取、浓缩、进样于一体的样品前处理技术,具有样品量小、萃取时间短、简便快捷、无需有机溶剂等诸多优点,尤其适合于大量样品的快速分析。根据吸附-解吸原理,由萃取纤维头吸附样品中的目标物,置于仪器进样口热解吸或溶剂溶解进入仪器进行分析。
固相微萃取通常有2种操作模式:顶空式和浸入式。前者适用于易挥发有机物的萃取,后者适用于大多数有机物的萃取[19]。敌百虫极性强,水中溶解度大,顶空式显然不适用。浸入式是富集水中敌百虫残留的可能途径。
商品化的萃取头仅涉及聚丙烯酸酯(PA)、聚二甲基硅氧烷(PDMS)、碳分子筛-聚二甲基硅氧烷(CAR-PDMS)、聚二甲基硅氧烷-二乙烯基苯(PDMS-DVB)和二乙烯基苯-碳分子筛-聚二甲基硅氧烷(DVB-CAR-PDMS)5种涂层。PA、PDMS纤维头富集分析物是基于吸收原理,而PDMS-DVB、CAR-PDMS和 DVB-CAR-PDMS纤维头是基于吸附原理[20]。基于吸附原理的萃取是竞争性过程,基于吸收原理的萃取则是非竞争过程[21]。另外,固相微萃取同样遵循“相似相溶”规则。PA纤维头具有极性涂层,适用于极性半挥发化合物的萃取[22];而 PDMS、PDMS-DVB、CARPDMS和DVB-CAR-PDMS纤维头的涂层基本为非极性,对某些极性物质也有富集作用。
由于敌百虫为极性半挥发物,故PA纤维头对其萃取效果更好。华勃等[23]使用85 μm PA纤维头浸入式萃取饮用水样品中的敌百虫等7种有机磷农药,得到的线性关系、检出限和精密度情况良好。
同样利用85 μm PA纤维头和浸入式固相微萃取模式,Adalberto等[24]对地表水和地下水中含敌百虫在内的16种农药组分进行了成功富集提取。
虽然固相微萃取法成功分析水中有机磷农残的报道很多,但涉及敌百虫组分的研究还比较少。限制其应用的因素主要有4方面:①可供选择的商品化萃取纤维头种类有限,且涂层厚度也有固定的规格,不能自由选取;②固相微萃取是基于吸附或吸收作用,对水中溶解度大的极性物质的富集作用可能有限;③纤维头价格较贵且属于消耗品,样品分析成本较高;④自动固相微萃取可保证萃取条件的一致性,但需要特殊装置来实现。
4 常用的仪器分析方法
4.1 气相色谱及气相色谱-质谱法
由于敌百虫的热稳定性差,在气相色谱分析过程中很容易发生分解,使得气相色谱及气相色谱-质谱仪直接用于敌百虫的测定受到了很大限制。
在受热条件下,敌百虫可以产生3种分解产物(敌敌畏、三氯乙醛和磷酸二甲酯)。其裂解方式有2种(图2):脱去氯化氢,重排生成敌敌畏;C-P键断裂,重排生成三氯乙醛和磷酸二甲酯。裂解方式以哪一种为主与色谱条件有关。

图2 敌百虫的2种裂解方式[25]
气相色谱的进样口温度、进样方式及柱箱升温速率均可影响敌百虫的降解,其中进样口温度是主要影响因素。进样口温度越低、柱箱升温速率越慢、进样速度越快,敌百虫的分解量就越少。另外,带有分流或不分流进样口的气相色谱分析敌百虫时,不论如何选择色谱条件,均存在敌百虫的分解现象[2]。冷柱头进样可避免敌百虫在进样口部位的热分解,但一般仪器标配不带有该进样口,需要另外选购。另外,由于敌敌畏是敌百虫的降解产物之一,因此气相色谱同时测定敌敌畏和敌百虫十分困难。
气相色谱及气相色谱-质谱仪直接分析敌百虫,理论上可采用2种定量方法:测定母体或测定分解产物。目前来看2种方法在实际操作中都存在一定问题。对于前者来说,敌百虫的热不稳定性始终影响测定结果的准确性;对于后者,敌百虫裂解方式的完整性和重现性不确定,以及样品中可能同时存在敌敌畏,都使得敌百虫的精确定量难以实现[26]。
配置常规进样口的气相色谱直接测定敌百虫有一定难度,衍生化的方法应运而生。一般来说,极性或热不稳定农药的分析需要先衍生化再进行气相色谱分析[18]。通过衍生反应,将敌百虫转化为热稳定性良好的其他物质后,便可使用气相色谱和气相色谱-质谱进行测定。甲基化、硅烷化、酰化[26-27]等衍生方法都已应用到了敌百虫的测定中。
使用冷柱头进样的气相色谱仪器条件可参考如下[27]:DB-1701(0.25 μm × 0.25 μm × 30 m),柱流量1.8 mL/min,进样量为1 μL。程序升温为80℃保持1 min,5℃/min到130℃,保持1 min,5℃/min到 190℃,20℃/min到 230℃,保持4 min。冷柱头温度为90℃保持1 min,40℃/min到230℃,保持25 min。检测器 NPD条件为250℃,铷珠电压20~30 pA,空气100 mL/min,氢气3.5 mL/min,尾吹(氦气)25 mL/min。
4.2 液相色谱及液相色谱-质谱法
液相色谱技术不受样品热稳定性的限制,非常适合热敏感物质的分析,一般常用的检测器为紫外和荧光检测器。
由于敌百虫的摩尔吸光系数较差,紫外检测器测定敌百虫的灵敏度很低[28]。同时,本身不具有荧光特性的敌百虫也不宜采用荧光检测器测定[29]。因此,液相色谱测定敌百虫一般也需要先进行化学衍生转化,得到具有较强紫外吸收或能发射荧光的物质。
利用敌百虫可催化联苯胺氧化反应的特性,通过测定联苯胺的氧化产物,Zhu等[28-30]建立了液相色谱-紫外检测器测定痕量敌百虫的方法。
在碱性条件下,敌百虫与特定的染料反应,生成具有荧光特性的离子对化合物。据此,EIKosasy[29]建立了敌百虫的荧光测定方法。
与液相色谱相比,质谱与液相色谱串联使用,灵敏度更高、选择性更好,不需衍生化步骤,可直接测定敌百虫。因此,在实际中应用也更为广泛[26,31-35]。
液相色谱-串联质谱法测定敌百虫的仪器条件可 参 考 如 下[33]:液 相 色 谱 条 件 为 C18柱(46 mm×50 mm),粒径1.8 μm;流动相为甲醇-水;梯度洗脱程序见表2;流速为200 μL/min;柱温为40℃;进样量为10 μL;离子源为电喷雾;扫描模式为正离子扫描;检测方式为多反应检测(MRM)。质谱条件为大气压电离(API);气帘气为206.85 kPa;雾化气为344.75 kPa;辅助加热气为55.00 L/min;碰撞气为34.47 kPa,离子源喷雾电压为5 000.00 V;离子源温度为450℃;检测离子对、去簇电压、碰撞能量见表3。

表2 流动相梯度洗脱程序

表3 检测离子对、去簇电压、碰撞能量[33]
5 结论
用于敌百虫测定的水样,采样前不要用样品荡洗玻璃采样瓶,水样应在pH<5.5的酸性条件下保存,并尽快萃取。
液-液萃取法间接提取敌百虫应严格控制反应条件。固相萃取法应选择对极性化合物保留能力良好的吸附剂富集敌百虫。固相微萃取的浸入模式可能是快速提取水中敌百虫残留的有效途径。
气相色谱、气相色谱-质谱仪及液相色谱分析敌百虫,都需要先衍生再测定。而质谱与液相色谱串联可直接测定敌百虫,在实际工作中应用更广。
[1]冯艳萍.手性有机磷农药对映体间联合毒性作用研究[D].浙江工业大学,2011:3.
[2]于慧娟,蔡友琼,沈晓盛,等.用气-质联用技术研究敌百虫在气相色谱分析过程中的分解产物[J].分析科学学报,2005,21(6):655-657.
[3]E-Pesticide Manual Version 5.0[DB].15th edition.British Crop Production Council,2010.
[4]Trichlorfon.Environmental Health Criteria 132[R].Geneva:World Health Organization,1992.
[5]GB 13192—1991 水质有机磷农药的测定 气相色谱法[S].
[6]HJ 493—2009 水质采样 样品的保存和管理技术规定[S].
[7]HJ/T 91—2002 地表水和污水监测技术规范[S].
[8]EPA 1657 The Determination of Organo-Phosphorus Pesticides in Municipal and Industrial Wastewater[S].
[9]ISO 5667-3 Guidanceon thePreservation and Handling of Water Samples[S].
[10]裘松.天然水的采样和样品保存[J].环境科学,1980(2):76-81.
[11]国家环境保护总局.水和废水监测分析方法[M].4版增补版.北京:中国环境科学出版社,2009:44.
[12]袁力.关于环境水质样品保存方法的讨论[J].环境研究与监测,2004,17(3):27-28.
[13]陈烨,谭丽,滕恩江,等.关于地表水中丙烯腈测定的样品保存条件[J].理化检验-化学分册,2013,49(4):483.
[14]牛喜业.水中敌百虫分析方法的研究[J].环境科学丛刊,1983,4(7):49-52.
[15]康跃惠,张干,盛国英,等.固相萃取法测定水源水中的有机磷农药[J].中国环境科学,2000,20(1):1-4.
[16]高智席,吴艳红,黎司,等.河水中噻菌灵和敌百虫的SPE-RP-HPLC 分析[J].中国农学通报,2012,28(6):217-221.
[17]冯复来,陆峰,向华.水中微量敌百虫直接测定方法的初步摸索——固相萃取法[J].净水技术,1998,64(2):25.
[18]Shamura Y,Tomiyama N,Murakoshi M,et al.Multi residue method of pesticides in water using automated solid phase extraction and liquid chromatographyatomspheric pressure chemical Ionization mass spectrometry[J].Journal of Pesticide Science,1998,23:241-249.
[19]徐溢,付钰洁.固相微萃取萃取头制备技术及试验方法的进展[J].色谱,2004,22(5):528-534.
[20]Supelco.CombipalSPME Option Manual[R].Pennsylvania:Sigma-Aldrich,2006.
[21]黄悯嘉,游静,梁冰,等.固相微萃取的涂层进展[J].色谱,2001,19(4):314-318.
[22]Supelco.Polyacrylate Film FiberforSolid Phase Microextracion of Polar Semivolatiles from Water[R].Pennsylvania:Sigma-Aldrich,1998.
[23]华勃,陈小辉,蚁焕钿.固相微萃取-气相色谱法测定生活饮用水中的有机磷农药[J].环境科技,2010,23(2):62-63.
[24]Adalberto M F,Fabio N S,Pereira P A.Development,validation and application of a method based on DISPME and GC-MS for determination of pesticides of different chemical groups in surface and groundwater samples[J].Microchemical Journal,2010,96(1):139-145.
[25]于慧娟,蔡友琼,李庆,等.气相色谱-质谱法研究敌百虫在气相色谱分析过程中产生的分解产物[J].色谱,2006,24(1):23-25.
[26]Maureen A N,Richard C.Determination of trichlorfon and dichlorvos residues in shrimp using gas chromatography with nitrogen-phosphorus detection[J].Journal of Agricultural and Food Chemistry,1996,44:2 686-2 689.
[27]Yamada N,Takahata J,Sasaki K,et al.Simultaneous determination of dichlorvos and trichlorfon in agricultural products by GC-FPD[J].食衛誌,2002,43(4):196-201.
[28]Zhu H Z,Liu W,Mao J W,et al.Cloud point extraction and determination of trace trichlorfon by high performance liquid chromatography with ultravioletdetection based on its catalytic effect on benzidine oxidizing[J].Analytica Chimica Acta,2008,614(1):58-62.
[29]EI-Kosasy A M. Fluorimetric determination of trichlorfon in pharmaceutical products in milk and in presence of its metabolites[J].Egyptian Journal of Pharmaceutical Sciences,2007,48:1-16.
[30]Zhu H Z,Cui Y M,Zheng X W,et al.Determination oftrace trichlorfon by high performance liquid chromatography with UV detection based on its catalytic effect on sodium perborate oxidizing benzidine[J].Analytica Chimica Acta,2007,548(1):166-171.
[31]张云,李耀平,李敏新,等.液相色谱-串联质谱法测定水产品中7种有机磷类农药残留物[J].分析实验室,2009,28(增刊):191-194.
[32]Wang G M ,Dai H,Li Y G,et al.Simultaneous determination of residues of trichlorfon and dichlorvos in animal tissues by LC-MS/MS[J].Food Additives and Contaminants,2010,27(7):983-988.
[33]李爱军,牟峻,王明泰,等.液相色谱-串联质谱法测定粮谷中敌百虫、辛硫磷残留量[J].农药,2010,49(8):599-601.
[34]黄捷,白桂昌,吕轶峰,等.快速高分离液相色谱-串联质谱法测定水产品中敌百虫残留[J].中国卫生检验杂志,2010,20(7):1 666-1 672.
[35]赵建晖,郑香平,吴文凡,等.液相色谱串联质谱法测定水产品中的敌百虫残留量[J].光谱实验室,2012,29(2):941-946.
猜你喜欢
江苏工程职业技术学院学报(2022年1期)2022-05-05 03:50:22
农业知识(2020年3期)2020-12-18 05:09:15
时代英语·高一(2019年5期)2019-09-03 02:09:34
食品与机械(2018年3期)2018-05-31 06:22:05
资源节约与环保(2018年1期)2018-02-08 02:18:26
中国塑料(2016年2期)2016-06-15 20:29:59
电测与仪表(2016年11期)2016-04-11 12:20:42
中国卫生标准管理(2015年17期)2016-01-20 09:26:42
电源技术(2015年5期)2015-08-22 11:18:28
乡村科技(2014年23期)2014-03-03 19:19:27
