
H2+体系中成键体系和反键体系能量与电子分布关系的研究
2015-04-07 05:53:40姚树文候振雨
河南科技学院学报(自然科学版) 2015年6期
姚树文,候振雨
(河南科技学院,河南新乡453003)
H2+体系中成键体系和反键体系能量与
电子分布关系的研究
姚树文,候振雨
(河南科技学院,河南新乡453003)
利用Hartree-Fock理论计算方法研究了H2+体系在不同键长的条件下,成键体系和反键体系的总能量、动能、势能随电子分布的规律.通过分析计算结果,发现成键体系总能量随第一个原子轨道系数a1的变化先减小后增大,动能随第一个原子轨道系数a1的变化先减小后增大,势能随第一个原子轨道系数a1的变化先增大后减小.无论是成键体系总能量还是反键体系总能量,都是受动能的影响更大.
成键体系;反键体系;总能量;动能;势能
星际空间中的大多数物质(暗物质、暗能量除外)几乎都处于等离子状态[1-2].中性原子在电离过程中,其原子核与核外电子完全分离,原子核彻底处于电子的包围中.许多物理过程都对离子体重的电子分布有重要影响,如高能辐射、光线偏振、弱相互作用等[2].化学反应过程中,也会有各种离子中间体出现,化合物在离子状态下的电子分布变化对理解其势能、动能、总能量以及其反应活性有重要的指导意义[3-7].
本文利用Gaussian 09软件计算H2+体系中成键体系和反键体系的势能、动能、总能量随着电子分布变化的规律.对于初学者有助于其理解化合物在离子状态下,电子分布的变化对于离子的势能、动能、总能量的影响,以及电子如何分布化合物的离子状态最稳定.
1 计算方法
运用Gaussian09软件计算不同键长下H-H间的轨道系数、哈密顿量和原子双电子积分,以及交换项、排斥项和FOCK矩阵元.设置氢、氢键长为0.6、0.8,Hartree-Fock(基组为STO-3G)方法,打印所有轨道系数.将不同键长时H-H间的轨道系数、哈密顿量和原子双电子积分与其键长一一对应起来.两种键长下依次设置某一氢原子的电子数a1,结合计算结果中的φ1φ2值,使用公式a12+a22+2a1a2φ1φ2=1计算得到另一氢原子的电子数a2,然后依据计算结果中的动能和势能项获得最终的成键轨道的动能、势能、总能量,以及反键轨道的动能、势能、总能量.最后对结果进行总结分析.
2 结果与分析
2.1 键长为0.06 nm时成键体系和反键体系数据
表1列出了键长为0.06 nm时,第一个原子上的电子数分别从1到0时,成键轨道的动能、势能和总能量.表2列出了键长为0.06 nm时,第一个原子上的电子数分别从1到0时,反键轨道的动能、势能和总能量.同时,分别将a1上的电子数与这3种能量作图,得到图1到图6.
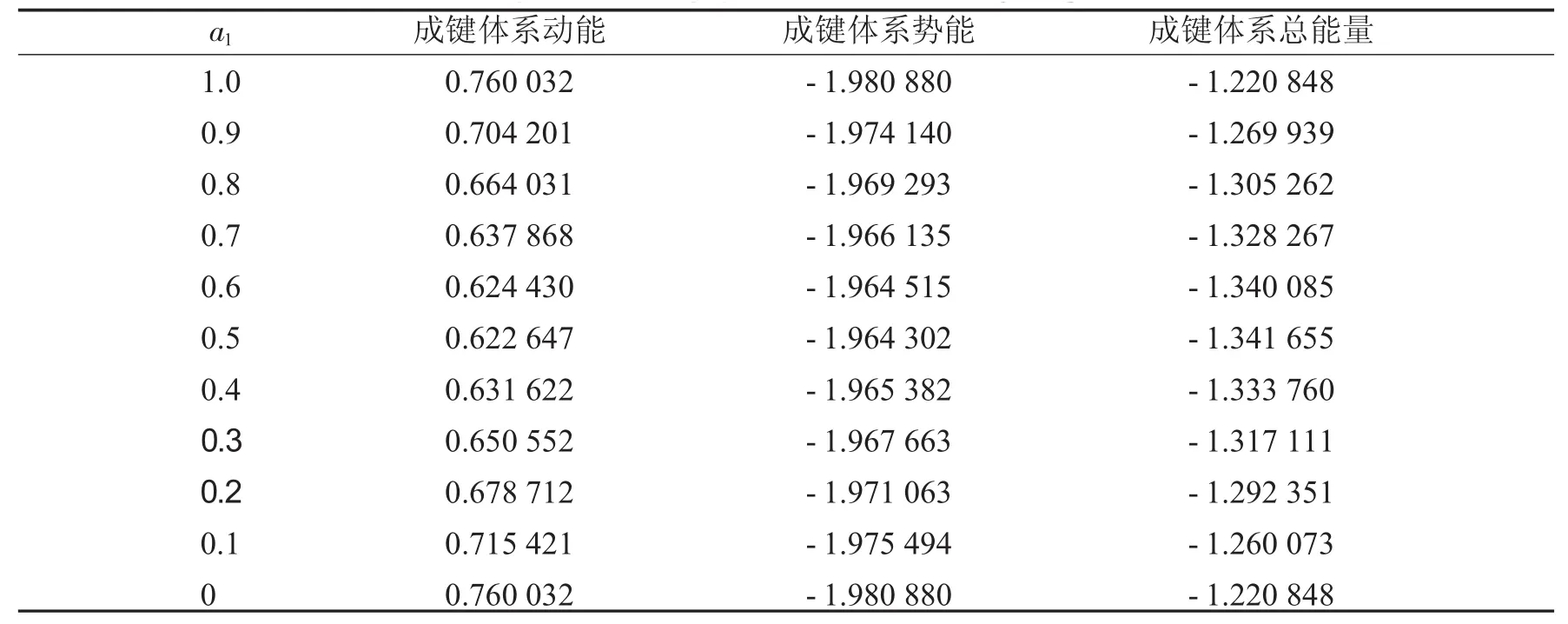
表1 键长=0.06 nm时成键体系能量Tab.1 The energies ofbondingsystems when bond length equals to0.06 nm
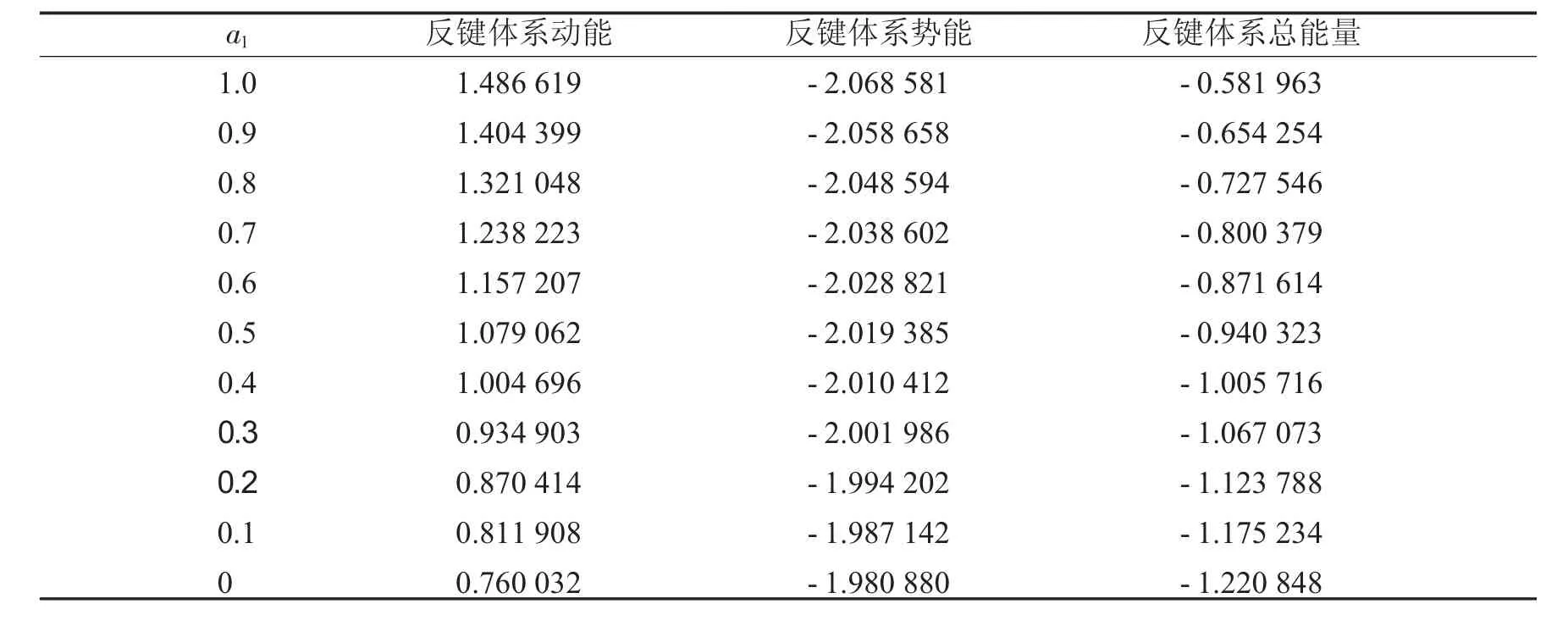
表2 键长=0.06 nm时反键体系能量Tab.2 The energies ofanti-bondingsystems when bond length equals to0.06 nm
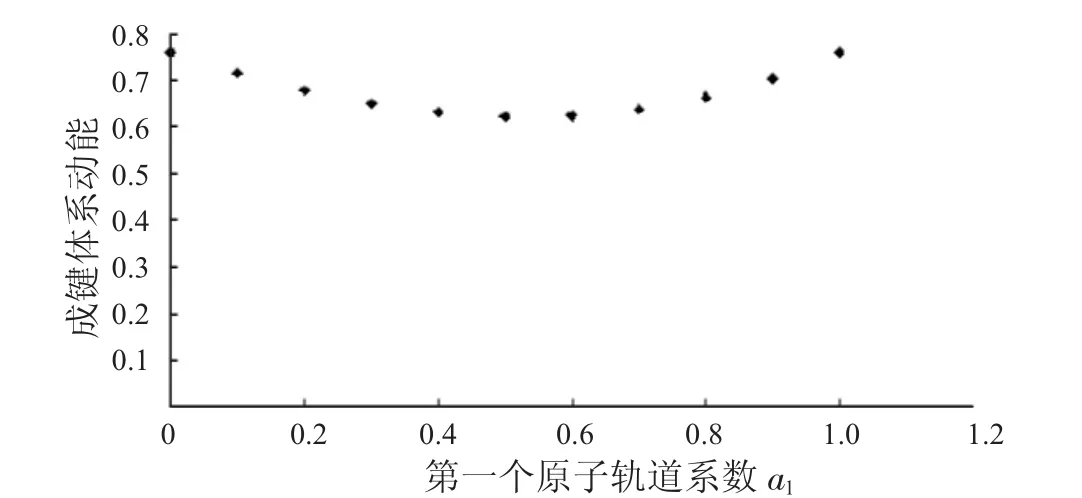
图1 成键体系动能与a1的关系Fig.1 The relationship between a1and kinetic energies ofbondingsystems
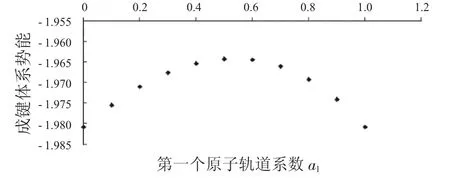
图2 成键体系势能与a1的关系Fig.2 The relationship between a1and potential energies ofbondingsystems
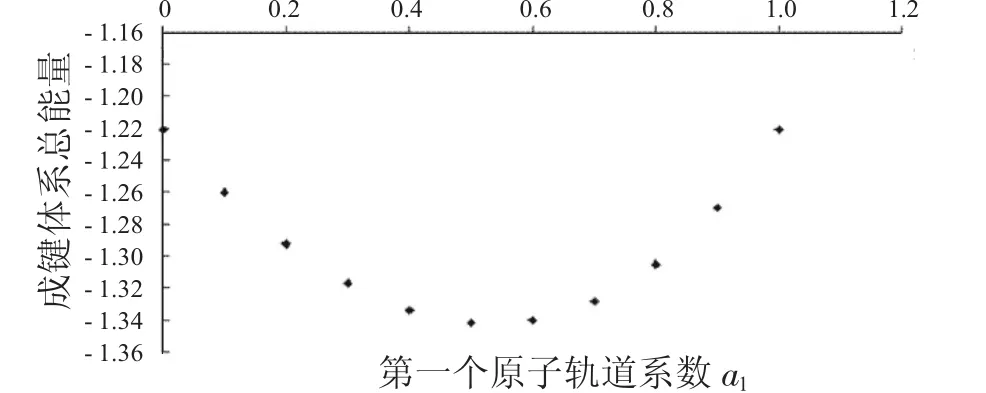
图3 成键体系总能量与a1的关系Fig.3 The relationship between a1and total energies ofbondingsystems
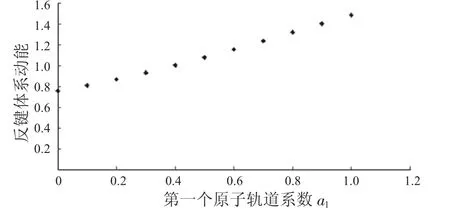
图4 反键体系动能与a1的关系Fig.4 The relationship between a1and kinetic energies ofanti-bondingsystems
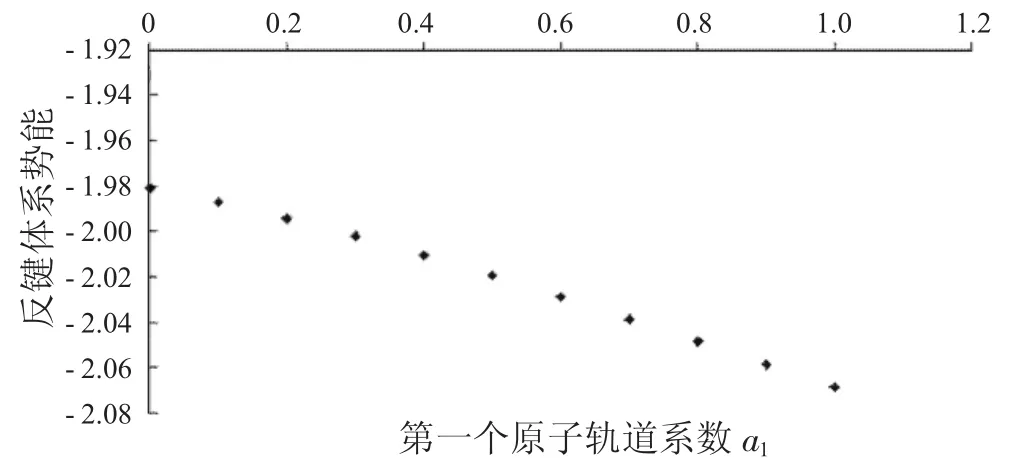
图5 反键体系势能与a1的关系Fig.5 The relationship between a1and potential energies ofanti-bondingsystems
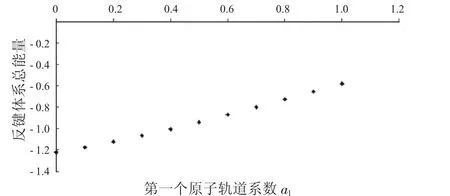
图6 反键体系总能量与a1的关系Fig.6 The relationship between a1and total energies ofanti-bondingsystems
从图1中可以看到随着第一个原子上的电子数从1减少到0,成键体系的动能逐渐减少,到一个最小值后,又重新增加.图2显示其势能则呈现先增加再减小的趋势.由于体系总能量等于体系势能与动能之和,而图3中显示的动能比势能的绝对值大很多,所以总能量的变化趋势与动能相同,呈现出先减小后增大的趋势.图4到图6中的反键体系呈现出类似的趋势.
2.2 键长为0.08 nm时成键体系和反键体系数据
表3列出了键长为0.08 nm时,第一个原子上的电子数分别从1到0时,成键轨道的动能、势能和总能量.表4列出了键长为0.08 nm时,第一个原子上的电子数分别从1到0时,反键轨道的动能、势能和总能量.同时,分别将a1上的电子数与这3种能量作图,得到图7到图12.
表3 键长为0.08 nm时成键体系能量Tab.3 The energies ofbondingsystems when bond length equals to0.08 nm
表4 键长为0.08 nm时反键体系能量Tab.3 The energies ofanti-bondingsystems when bond length equals to0.08 nm
图7 成键体系动能与a1的关系Fig.7 The relationship between a1and kinetic energies ofbondingsystems
图8 成键体系势能与a1的关系Fig.8 The relationship between a1and potential energies ofbondingsystems
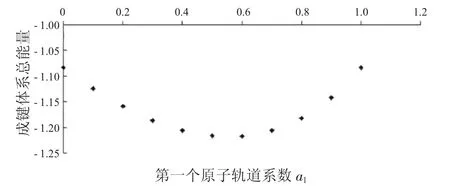
图9 成键体系总能量与a1的关系Fig.9 The relationship between a1and total energies ofbondingsystems
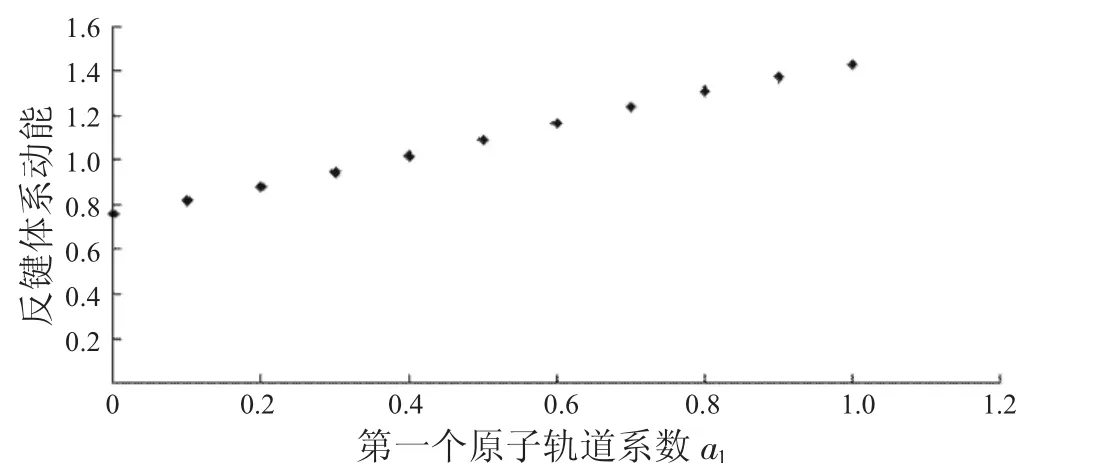
图10 反键体系动能与a1的关系Fig.10 The relationship between a1and kinetic energies ofanti-bondingsystems
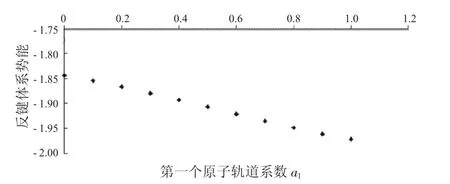
图11 反键体系势能与a1的关系Fig.11 The relationship between a and potential energies ofanti-bondingsystems
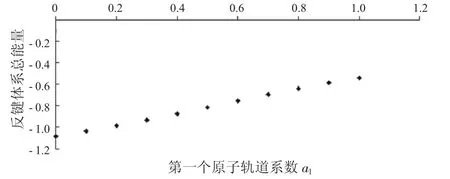
图12 反键体系总能量与a1的关系Fig.12 The relationship between a1and total energies ofanti-bondingsystems
从图7中可以看到随着第一个原子上的电子数从1减少到0,成键体系的动能逐渐减少,到一个最小值后,又重新增加.从图8中可以看出,其势能则呈现先增加再减小的趋势.由于体系总能量等于体系势能与动能之和,而动能比势能的绝对值大很多,所以在图9中总能量的变化趋势与动能相同,呈现出先减小后增大的趋势.从图10到图12中可以看出,反键体系的这3种能量也有类似的结果.整体结果对于理解不同电子分布对氢分子离子的能量影响有一定参考意义.
3 小结
成键体系总能量随第一个原子轨道系数a1的增大先减小后增大,动能随第一个原子轨道系数a1的增大先减小后增大,势能随第一个原子轨道系数a1的增大先增大后减小.反键体系总能量随第一个原子轨道系数a1的增大而增大,动能随第一个原子轨道系数a1的增大而增大,势能随第一个原子轨道系数a1的增大而减小.无论成键体系总能量还是反键体系总能量都是受动能的影响更大.
[1]徐仁新.天体物理导论[M].北京:北京大学出版社,2006.
[2]尤峻汉.天体物理中的辐射机制[M].2版.北京:科学出版社,1996.
[3]Shea J A.Flygare W H.The rotational spectrum and molecular structure of the ethylene-HF complex[J].J.Chem.Phys., 1982,76(10):4857-4865.
[4]Kirschner KN,Woods R J.Quantummechanical studyofthe nonbonded forces in Water-Methanol complexes[J].J Phys ChemA, 2001,105(16):4150-4155.
[5]BaderR FW.AQuantumTheory[M].Oxford:Oxford University,1990.
[6]Frisch MJ,Trucks GW,Schlegel H B,et al.Gaussian 09[CP/OL].Pittsburgh,PA:Gaussian,Inc.,2005.
[7]Watts J D,FranciscoJ S.Ground and electronicallyexcited states ofmethyl hydroperoxide:Comparison with hydrogen peroxide[J]. J.Chem.Phys.,2006,125(10):104301-104309.
(责任编辑:卢奇)
A study on the effect of electron distribution on the energy of H2+
YAO Shuwen,HOU Zhenyu
(Henan Institute ofScience and Technology,Xinxiang453003,China)
The total energy,kinetic energy,potential energy of the bonding and anti-bonding system at different bond were studied with Hartree-Fock method.The results showed that the total energy firstly increases and then decreased with the electron number on the first atom,the kinetic energy had the same trend as the total energy,but the potential energy had an opposite trend to the total energy.The total energy was mainly determined by the kinetic energy not only for the bonding system but also for the anti-bonding system.
bonding system;anti-bonding system;total energy;kinetic energy;potential energy
TN911.72
A
1008-7516(2015)06-0047-06
10.3969/j.issn.1008-7516.2015.06.009
2015-10-26
河南省科技厅基础与前沿项目(102300410128)
姚树文(1963-),河南辉县人,博士,副教授.主要从事计算化学研究.
猜你喜欢
吉林大学学报(理学版)(2025年1期)2025-02-06 00:00:00
高中数理化(2023年6期)2023-08-26 13:28:24
中学生数理化·八年级物理人教版(2023年6期)2023-05-25 11:59:48
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
——《势能》
文化纵横(2022年3期)2022-09-07 11:43:18
中学生数理化·八年级物理人教版(2022年6期)2022-06-05 06:55:40
中学生数理化·八年级物理人教版(2021年6期)2021-11-22 07:49:52
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
考试周刊(2018年39期)2018-04-19 10:39:44
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
