
非均相催化臭氧氧化深度处理炼油废水
2015-03-19 01:57邓凤霞岳秀丽徐善文
浙江大学学报(工学版) 2015年3期
邓凤霞,邱 珊,岳秀丽,徐善文,陈 聪,丁 晓,马 放,
(1.哈尔滨工业大学 市政环境工程学院,黑龙江 哈尔滨150090;
2.哈尔滨工业大学 城市水资源与水环境国家重点实验室,黑龙江 哈尔滨150090)
委内瑞拉超重原油属于高密度、高含硫、高氮、高残炭、高金属、高酸值的非常规原油[1].加工这种原油所排出的污水污染物含量高,含盐量高,可生化性差.废水中的污染物主要包括石油烃类、硫化物、挥发酚、悬浮物、氨氮以及其他有毒金属物质.废水的COD较高,难降解物质较多,而且,受碱性水和酸性水的影响,废水的p H值变化较大[2].根据国家节能减排的发展战略与石化企业的节水要求,石化企业不仅要持续减少污染物的排放,还需节约水资源,提高水的循环使用率,必须对出水进行深度处理[3].臭氧氧化作为一种实用、高效的高级氧化技术,具有氧化能力强、反应时间短、无二次污染、设备简单等优点.在某些条件下,臭氧在反应过程中会产生氧化能力更强的羟基自由基·OH.它是目前用于水处理的最强氧化剂,是出水深度处理的最佳选择[4].2条公认的臭氧氧化机理如下[5]:一是臭氧的直接氧化,有选择性,反应的速率常数为100~103mol-1·L·s-1,易氧化含双键、苯环结构、胺等有机物;二是臭氧的间接氧化,主要是通过产生自由基来完成,无选择性,其反应速率常数大得多,一般108~1010mol-1·L·s-1.在酸性条件下,以臭氧的直接氧化为主.在碱性溶液中将发生如下链式反应[6]:

本文以经过“隔油—气浮—生化”传统老三套为主体工艺处理的委内瑞拉超重原油废水生化出水作为研究对象,采用自主研究的铜锰氧化物为催化剂,进行非均相催化臭氧氧化实验,探讨非均相催化臭氧氧化工艺对废水的深度处理效能,探索最佳影响参数,并对其机制进行了初步探索.
1 实验材料与方法
1.1 废水来源与水质
废水取自广东某炼油企业委内瑞拉超重原油废水生化处理后的出水.水质为:ρ(COD)=87~114 mg/L,p H=6.8,色度为45,石油类3 mg/L,氨氮为10 mg/L,来水随生产有一定波动性.处理目标是使出水污染物浓度满足该炼油企业的回用水标准:ρ(COD)<50 mg/L.
1.2 实验装置
在静态实验过程中,以纯氧作为臭氧发生器气源,采用钛板微孔曝气加强臭氧的传质作用.臭氧接触反应器内径为50 mm,高为1 m,有效容积为1 L.反应装置见图1.
试验前先用纯水冲洗反应器,再用臭氧预氧化10 min去除反应器中可能消耗臭氧的成分,然后排空反应器中的气体,并用蒸馏水冲洗2次.准备水样500 m L,按实验所需加入不同质量浓度的催化剂.将反应溶液加入臭氧接触反应器中,打开臭氧发生器,调节气体流量计,控制不同臭氧投加的质量浓度.开始试验计时,在不同时刻取样,取出的水样用氮气吹脱3 min终止反应,水样经过静沉后,用于测定所需指标.
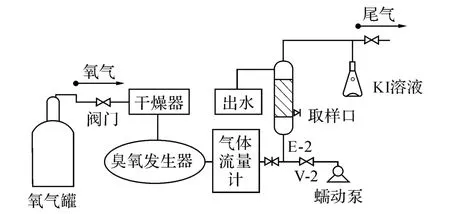
图1 臭氧催化氧化实验装置图Fig.1 Experimental setup for catalytic ozonation
1.3 分析指标与方法
分析指标COD、p H值、臭氧质量浓度和催化剂中Cu2+、Mn2+质量浓度分别采用重铬酸钾法、玻璃电极法、碘量法[7]和原子吸收分光光度法测定.采用电子万能材料试验机(Instron 5500R)[8]分析催化剂抗压强度.反应前后的水质用GC-MS(Agilent 6890GC/5973MSD)进行成分分析,采用30 m×0.25 mm×0.25μm的HP5MS毛细柱,分流进样模式,分流比为1:20.以高纯氮气作为载气,流速为1.1 m L/min,程序升温,初始温度为60℃,保持3 min,以25℃/min的速度进行升温,上升到320℃.接口和进样口温度均为270℃[9].
1.4 催化剂制备及其特性
实验使用的催化剂是自主研制的铜锰氧化物催化剂.制备方法如下:以粒径为2~6 mm的Al2O3作为催化剂载体,进行活化.选用Mn、Cu的硝酸盐作为前驱物质,配成溶液后,采用等体积浸渍法将活性组分负载于活化后的载体上进行加工;然后在140℃下烘干.24 h后在600℃下焙烧5 h,即可得到颗粒状铜锰氧化物催化剂CuOχ-MnOχ/Al2O3.
分别采用Zeta电位法测量催化剂等电点[10],通过BET比表面积测试法、SEM (scanning electron microscope)、XRD (X-ray diffraction)表征催化剂表面特性.催化剂的等电点为p H=6.780,BET比表面积为184.060 m2/g,孔径d=8.34 nm,属于介孔材料,其吸附脱附曲线及孔径分布曲线如图2所示,其中va为每克催化剂所吸附的氢气体积,vh为每克催化剂的孔体积.SEM扫描结果显示催化剂表面空隙发达,如图3所示.如图4所示的XRD结果显示该催化剂为非晶态,2θ为扫描角度.
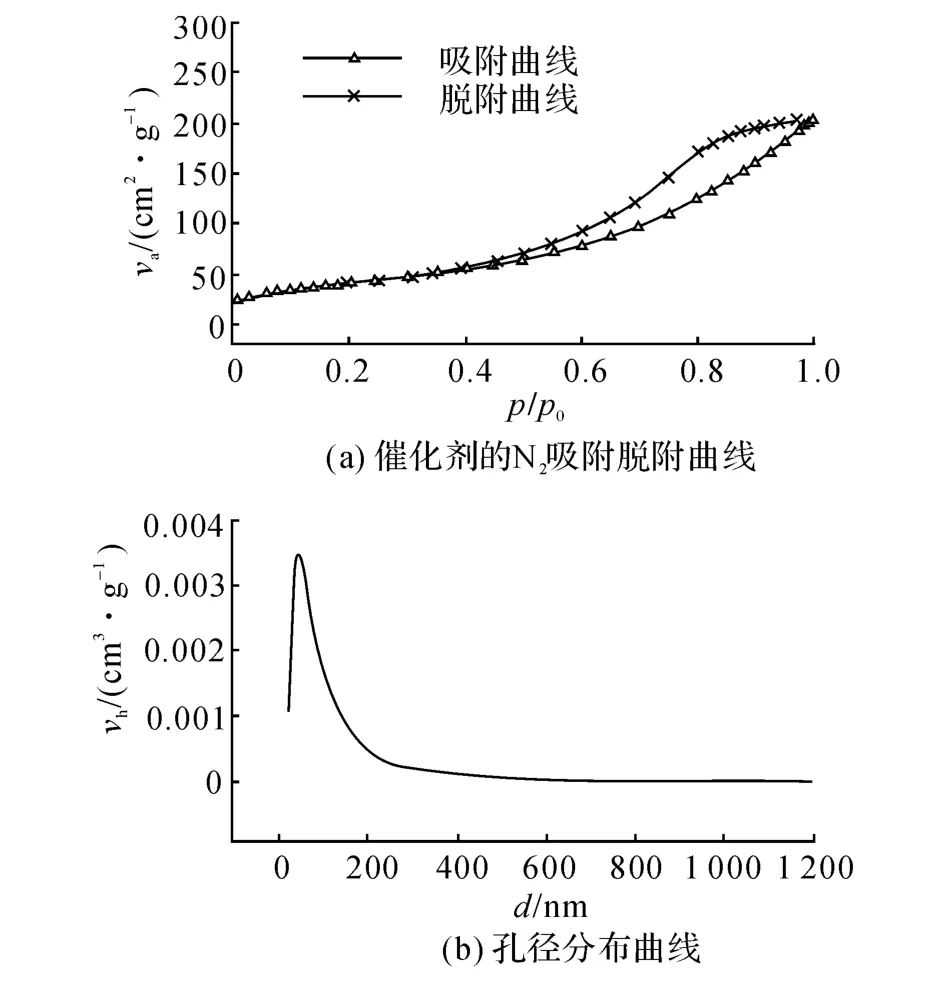
图2 催化剂的N2吸附脱附曲线和孔径分布曲线Fig.2 N2 adsorption-desorption isoth and pore size distribution of catalyst
2 结果及分析
2.1 p H值的影响
p H值是确定臭氧催化氧化是以臭氧直接氧化为主还是以自由基氧化为主的关键因素.溶液p H值影响催化剂的表面性质,从而影响非均相催化臭氧氧化的效果.催化剂质量浓度为3 g/L,臭氧投加质量浓度为50 mg/L,室温(17℃)条件下,考察不同p H值对催化效果的影响.如图5(a)所示,反应前5 min,COD去除率明显增加,10 min后,COD去除率增加趋势减缓.碱性条件下的COD去除率高于酸性条件,并且在中性条件下,COD去除率最高,达到98.875%.分析可知,非均相催化臭氧氧化具有最佳的p H值(p H=7),满足该炼油企业的回用水标准.考虑到原水的p H=6.8(接近7),所以选原水的p H值作为最佳反应p H值.如表1所示,k为表观速率常数,对比不同p H值下COD降解的动力学,通过ln(C/C0)对t作图可知:不同p H 值下的ln(C/C0)与t呈线性关系,反应符合一级动力学关系.当p H值为7时,反应速率常速为最大,即3.120 s-1.

图3 不同放大倍数催化剂的SEM图Fig.3 The SEM images of the catalyst under different magnifications

图4 催化剂的XRD图Fig.4 The XRD pattern of the catalyst
金属氧化物催化降解水中物质的能力取决于金属氧化物表面的性质.由于金属氧化物表面的金属离子配位一般不饱和,在水中金属氧化物表面会吸附水,离解生成氢离子和氢氧根离子,从而形成表面羟基[11].由于金属氧化物表面吸附的羟基数目,电荷状态亦不同,可用金属表面的p Hzpc(p Hpzc即等电点,是指金属氧化物表面静电荷为0时的p H值)来表示,p Hzpc与溶液的p H值有关,即
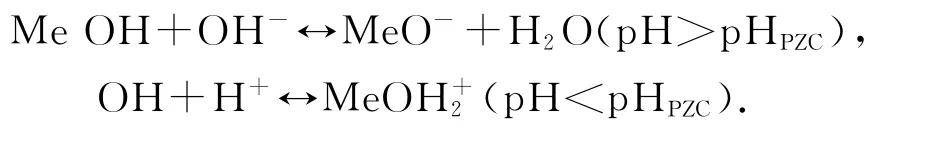
所采用的催化剂由Al2O3,CuO及MnO2组成,且等电点为6.78.陈忠林等[12]以γ-Al2O3粉末为催化剂,研究催化臭氧氧化去除水体中典型嗅味物质2-甲基异茨醇(MIB)的效能与机理,当催化剂表面的p Hpzc和溶液p H值近似相等时,催化剂的催化活性最高.与实验中p H值为7时催化效率最高的结论一致.在过强酸性条件下,产生的少量自由基不能稳定存在[13],导致p H=1时,COD去除率明显偏低.随着p H值的提高,出现自由基引发剂OH-,加速自由基的产生[14],COD去除率明显升高.当碱性进一步增加时,自由基相互碰撞的几率增加,引起自由基自身的猝灭效应,同时高浓度自由基会成为体系自由基捕捉剂,从而导致自由基链式反应传递受阻[15].
对不同p H值反应过程中的p H值变化进行监测.从图5(c)中可以看出:在前20 min,p H值呈现下降趋势,而20 min后p H值小幅度升高.从后文研究的GC-MS可知,废水经过臭氧的催化氧化,大分子有机物被氧化为小分子有机酸,这是p H值下降的主要原因.随着小分子有机酸被矿化为CO2和H2O,体系的p H值小幅度升高[16].

表1 不同p H值对表观速率常数的影响Tab.1 Effect of different p H values on apparent reaction______rate constant
2.2 不同催化剂质量浓度的影响
选择合适的催化剂是节约成本的重要途径.臭氧投加质量浓度为50 mg/L,p H=6.8(原水),室温(17℃)条件下考察不同催化剂质量浓度对非均相催化臭氧氧化去除COD的影响.从图6(a)可得:催化剂能明显提高COD的去除率,并且随着催化剂质量浓度的增加,催化臭氧氧化对COD的去除率越来越高.当催化剂质量浓度低于3 g/L时,随着催化剂质量浓度的增加,COD去除率增加的幅度较明显,当催化剂质量浓度高于3 g/L时,增加催化剂质量浓度对于提高COD去除率效果不明显.所以,从技术可行性和经济合理性角度考虑,以3 g/L作为本实验的最适宜催化剂质量浓度.在不同催化剂质量浓度下,COD的ln(C/C0)对t作图,符合一级动力学反应,表明随着催化剂质量浓度增加,反应速率也随之增快.

图5 不同p H值对COD的去除率影响Fig.5 Effectof different p Hvalues on CODdegradation
大量研究表明[17]:非均相催化臭氧氧化与催化剂表面的活性位点数目有关.增加反应体系中催化剂的投加质量浓度,一方面,会增加催化剂的活性位点数目,增大臭氧、污染物与催化剂表面的接触几率.这有利于臭氧和污染物质在催化剂表面的物理和化学吸附[18],促进后续反应.另一方面,随着催化剂投加质量浓度的增加,催化剂在臭氧气体的扰动下,相互碰撞的几率增加,导致催化剂中的活性组分Mn2+产生少量的溶出,将会发生[19],从而促进羟基自由基的产生,进一步加强COD的去除(X为有机物).


图6 不同催化剂质量浓度对COD却除率和表观速率常数的影响Fig.6 Effect of different catalyst dosages on COD degradation and apparent reaction rate constant

表2 不同催化剂质量浓度对表观速率常数的影响Tab.2 Effect of different catalyst dosages on apparent reaction rate constant
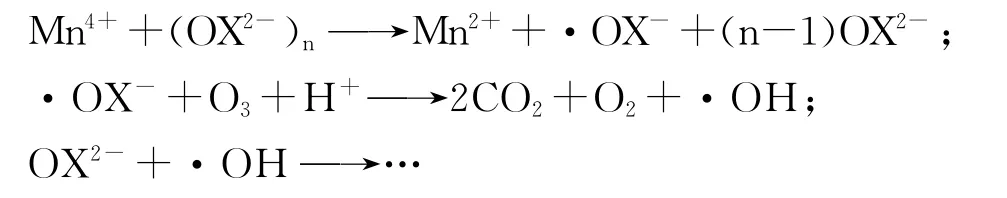
2.3 温度的影响
由阿伦尼乌斯公式可知,升高温度能显著加快化学反应的速率.研究不同温度对非均相催化臭氧氧化的影响,有利于机理的探讨.在臭氧投加质量浓度为50 mg/L,p H=6.8(原水),催化剂质量浓度为3 g/L的情况下,考察不同温度对非均相催化臭氧氧化去除COD的影响.由图7可知:当体系温度提高时,反应速率加快,可见温度对COD的去除率影响显著.温度从10℃上升到20℃时的COD去除率高于温度从20℃上升到50℃时的COD去除率.图7(b)是ln(C/C0)对反应时间t作图,反应体系中臭氧质量浓度过量,从动力学角度认为臭氧质量浓度是常数.可以看出,ln(C/C0)对t具有良好的线性关系,符合一级动力学方程.从表3可以看出,当温度t0从10℃升高到50℃时,k从0.024 min上升到0.082 min,提高了3.4倍.随着温度的升高,表观速率常数增大,这有利于提高COD的去除率.从表3可以看出,lnk与1/T具有良好的线性关系,将lnk对1/T进行拟合,方程为y=-3 118.400χ-7.428,R2=0.958,根据Arrennius公式,可以求出该催化氧化反应的活化能为25.926,低于臭氧氧化体系的活化能(60.145),说明非均相催化臭氧氧化有利于降低反应体系的活化能,加快COD的去除.

图7 反应过程中p H值变化Fig.7 The p H value changes during reaction

表3 不同温度对表观速率常数的影响Tab.3 Effect of different temperatures on the apparent reaction rate constant
反应体系温度从10℃升高到50℃时,COD的去除率整体呈现上升趋势.然而,随着温度的升高,臭氧的溶解度发生急剧变化.10℃时臭氧在水中的溶解度为0.780 g/L,而温度上升到50℃时,溶解度仅为0.190 g/L[20],因此,高温溶液中臭氧浓度低,臭氧催化氧化的反应速率降低.另一方面,温度升高使亨利常数增加,减小了气相臭氧进入液相的传质推动力[21].在温度升高的过程中,高温下COD去除率的上升幅度小于低温下COD去除率的上升幅度.鉴于使废水升温不经济的事实,选择在室温条件下进行实验.
2.4 臭氧投加质量浓度的影响
合适的臭氧投加质量浓度不仅能够满足技术要求,而且能够达到经济利益最大化.本实验探究不同臭氧投加质量浓度对催化效果的影响.通过调节氧气流质量浓度和臭氧发生器上不同功率的调节旋钮来调节臭氧产量.图8(a)为p H=6.8,催化剂用质量浓度为3 g/L,室温(17℃)条件下,不同臭氧投加质量浓度对COD去除率的影响.随着臭氧投加质量浓度的增加,COD去除率逐渐升高.当臭氧投加质量浓度从30 mg/L上升到60 mg/L时,COD去除率从60 mg/L上升到80 mg/L.臭氧投加质量浓度与COD去除率并非呈线性增长,结果显示:单纯依靠提高臭氧投加质量浓度来提高COD降解效率并不经济,应综合考虑经济和效率来确定合适的臭氧投加质量浓度,本实验选择最适宜臭氧投加质量浓度为50 mg/L.将不同臭氧质量浓度下COD的ln(C/C0)对t作图,如图8(b)所示.可以看出,ln(C/C0)对t具有良好的线性关系,符合一级动力学方程.且臭氧质量浓度从30 mg/L增加到80 mg/L时,k增加了2.7倍,如表4所示.

表4 不同臭氧投加质量浓度对表观速率常数的影响Tab.4 Effect of different ozone dosages on the apparent re-______action rate constant
通过改变气体流量控制臭氧投加质量浓度,传质过程的控制步骤为臭氧从气相到液相的过程,该过程受诸多因素的影响.从图8(a)可以看出,随着通入气量的增加,臭氧的气相浓度降低,气液界面上扰动加剧,一定程度上减少了传质过程的气膜阻力,同时增大了气液接触面积.最终COD去除率的提高得益于传质速率的提高.
2.5 催化剂的稳定性

图8 不同臭氧浓度对COD去除率和表观速率常数的影响Fig.8 Effectof different catalyst dosages on COD degradation and apparant reaction rate constant
催化剂重复利用的稳定性是该技术能实现工业化的一个重要指标.为了考察催化剂的重复使用效果,在上述实验确定的最佳条件下,即:p H值为原水p H=6.8,催化剂用质量浓度为3 g/L,臭氧投加的质量浓度为50 mg/L,室温(17℃)条件下,进行重复试验20次,每次反应15 min,考察催化剂的抗压强度,活性组分Cu2+、Mn2+的流失情况及非均相催化氧化对COD去除率的影响,见表5、6,图9(a)及(b).
由表5可得,在反应20次后,催化剂的最大压缩载荷Fmax变化仅为13.467N,反应前后抗压性能稍有变化.而从表6可得,作为活性组分的Cu2+、Mn2+在反应20次后变化分别为0.001%和0.124%,而载体的主要成分Al同样在反应20次后仅仅损失了0.570%.这也验证了以上结论:在反应20次后,该催化剂对COD去除率的作用效果基本不变.说明了在反应前后,催化剂的抗压能力,活性组分含量未发生变化,催化效果稳定,未出现催化剂失活现象.同时在催化剂反应20次后,检测水中锰离子和铜离子含量,以确定金属流失量.其中Mn2+和Cu2+分别为0.015 mg/L和0.059 mg/L.金属流失少,满足《GB/T19923-2005》中对 Mn2+< 0.1 mg/L的要求,不会造成二次污染.

表5 不同反应次数后催化剂的最大压缩载荷Tab.5 Maximum load of catalyst after different reaction times

表6 不同反应次数后催化剂活性组分质量分数Tab.6 Active component contentof catalyst after different reaction times
从图9(a)中可以看出,在最适宜条件下装置间歇运行20次,进水COD为87~114 mg/L,出水COD 为40.100~50.400 mg/L,COD 去除率为43.590%~63.110%.COD符合该炼油企业的回用水标准.也能在一定程度上去除氨氮,见图9(b).同时,催化剂在反应20次后,对于COD去除率保持稳定状态,进一步证明反应前后催化剂的性能保持稳定.
利用非均相催化臭氧氧化技术深度处理炼油废水,在p H=6.8,催化剂质量浓度为3 g/L,臭氧投加质量浓度50 mg/L,室温(17℃)条件下,反应15 min后,出水符合该炼油企业的回用水标准.按产生每克臭氧需电耗15 W计算,吨水处理耗电量为1.2 k W,按电费0.7元/度计,非均相催化臭氧氧化电费为0.84元/t.

图9 最佳条件下的COD及氨氮去除效果Fig.9 COD degradation and ammonia nitrogen removal effect under optimum conditions
2.6 臭氧催化臭氧氧化机制初探
为初步探究非均相催化臭氧氧化机制,将反应前后的水样进行GC-MS分析对比,考察非均相催化臭氧氧化工艺对炼油废水中有机物的去除效能.反应前后的总离子流见图10.有机物的质量浓度用峰面积近似表示,结果见表7.在GC-MS分析中,水中有机物的出峰时间顺序与其分子沸点、分子质量由低到高的排列顺序一致.所以,保留时间(t′)低的色谱峰代表有机物分子沸点、质量均低[22].因此,将GC-MS检出的有机物按出峰的保留时间进一步分类整理,根据臭氧催化氧化处理前后水样中有机物的保留时间分布变化,进一步了解有机物结构和分子质量的变化规律结果(如图11所示).由表7可得,生化出水检测出119种物质,包括烷烃、烯烃、醇、酯及其他含氮、硫的杂环有机物等,其分子量大,化学结构复杂.经过臭氧催化氧化工艺处理后,有机物降为81种,其中新增了4种有机酸和酯以及3种醛和酮,而其他含氮、硫的杂环有机物减少了45种.这说明臭氧催化氧化工艺对化学结构复杂的大分子有机物去除效果明显.臭氧的直接氧化通过环加成机理,即臭氧的偶极距结构与不饱和键有机物加成导致键断裂,从而生成相应的醛、酮、酯和酸,这是导致废水经过臭氧催化氧化后有机酸、酯、醛和酮增加的主要原因.如图11所示,经过臭氧催化氧化处理后,废水有机化合物的结构发生显著变化.臭氧催化氧化前,废水中保留时间为20~30 min是分子质量大、沸点高的有机物,并且其质量分数占68.070%,其中主要为含氮和硫的杂环化合物.

表7 GC-MS分析反应前后峰个数和峰面积Tab.7 Peak number and peak area before and after reaction______by GC-MS analysis

图10 GC-MS分析反应前后的总离子流图Fig.10 Total ion flow diagram before and after reaction by GC-MS

图11 反映前后出水不同保留时间内有机物分布Fig.11 Distribution of organic matter in different retention time before and after reaction
而经过臭氧催化氧化工艺后,保留时间为20~30 min的有机物质量分数下降了78.240%,其中保留时间为10~20 min的氧化产物最多,占56.780%,主要为有机酸和酯类化合物.这说明臭氧催化氧化能将复杂大分子、高沸点有机物质氧化为简单小分子物质.同时,检测反应前后水质的B/C从0.106提高到0.289,说明臭氧氧化可以提高水质的可生化性.
3 结 论
(1)非均相催化臭氧氧化工艺用于深度处理炼油废水可行,采用自主研究的铜锰氧化物催化剂及非均相催化氧化反应器对炼油废水进行处理,对COD去除效果好.在室温为17℃,投加臭氧质量浓度为50 mg/L,p H值为6.8,催化剂投加质量浓度为3 g/L时,进水COD的质量浓度为87~114 mg/L,出水COD的质量浓度为40.100~50.400 mg/L,符合该炼油企业回用水标准.同时对氨氮也有去除效果.
(2)自主研制的铜锰氧化物催化剂催化效果稳定,经多次使用,反应前后催化剂的抗压性能与活性组分未发生明显变化.催化剂未出现失活现象,催化剂可重复使用.
(3)非均相催化氧化工艺可以降解废水中的大分子有机物质,尤其是含氮的杂环物质,能将高沸点大分子有机物降解为低沸点小分子有机物.
(
):
[1]张抗,卢雪梅.委内瑞拉石油工业形势及对我国的影响[J].中外能源,2011,16(10):18- 36.ZHANG Kang,LU Xue-mei.The influence of venezuela's oil industry and its situation[J].Sino-Global Energy,2011,16(10):18- 36.
[2]孙武,王泉,王能才,等.国产PVDF中空纤维膜在炼油废水深度处理回用中的应用[J].石油炼制与化工,2013,44(03):79- 83.SUN Wu,WANG Quan,WANG Neng-cai,et al.The application of homemade PVDF in the advanced treatmentof oil refining[J].Petroleum Processing and Petrochemicals,2013,44(03):79- 82.
[3]刘春平.石油化工企业的节水减排[J].化工环保,2006,26(01):74- 77.LIU Chun-ping.Water saving and wastewater reducing in petrochemical industry[J].Environmental Protection of Chemical Industry,2006,26(01):74- 77.
[4]ZHAO L,MA W,MA J,et al.Characteristic mechanism of ceramic honeycomb catalytic ozonation enhanced by ultrasound with triple frequencies for the degradation of nitrobenzene in aqueous solution[J].Ultrasonics Sonochemistry,2014,21(1):104- 112.
[5]CAO H B ,XING L L,WU G G.Promoting effect of nitration modification on activated carbon in the catalytic ozonation of oxalic acid[J].Applied Catalysis B:Environmental,2014,146:169- 176.
[6]NIE Y L,HU C,LI N N.Inhibition of bromate formation by surface reduction in catalytic ozonation of organic pollutants overβ-FeOOH/Al2O3[J].Applied Catalysis B:Environmental,2014,147:287- 292.
[7]贾瑞平,盛敏奇,张辉,等.臭氧分析方法的研究和进展[J].工业水处理,2008,28(02):1- 5.JIA Rui-ping,SHENG Min-qi,ZHANG Hui,et al.Research and development of the analytical methods of ozone[J].Industrial Water Treatment,2008,28(02):1- 5.
[8]孙志忠.臭氧/多相催化氧化去除水中有机污染物效能与机理[D].哈尔滨:哈尔滨工业大学,2006.SUN Zhi-zhong.Efficiency and mechanism of ozone/heterogeneous catalytic oxidation for the degradation of organic pollutants in water[D].Harbin:Harbin Institute of Technology,2006.
[9]陈峰,唐访良,张明,等.废水中松节油类化合物的分析方法[J].中国给水排水,2013,29(10):86- 90.CHEN Feng,TANG Fang-liang,ZHANG Ming,et al.Determination of Turpentine-like Compounds in Wastewater[J].China Water and Wastewater,2013,29(10):86- 90.
[10]丘永梁,陈洪龄,汪效祖,等.水热法制备纳米TiO2及其等电点的研究[J].高校化学工程学报,2005,19(01):129- 133.QIU Yong-liang,CHEN Hong-ling,WANG Xiao-zu,et al.Preparation of nanometer titanium dioxide powders by hydrothermal method and their isoelectric points[J].Journal of ChemicalEngineering of Chinese Universities,2005,19(01):129- 133.
[11]W.斯塔姆编,汤鸿霄译.水化学[M].北京:科学出版社,1987:460- 465.
[12]陈忠林,齐飞,徐冰冰,等.γ-Al2O3催化臭氧氧化水中嗅味 物 质 机 理 探 讨 [J].环 境 科 学,2007,28(3):563- 568.CHEN Zhong-lin,QI Fei,XU Bing-bing,et al.Mechanism discussion ofγ-alumina catalyzed ozonation for 2-Methylisoborneol removal[J].Enviromental Science,2007,28(3):563- 568.
[13] NAWROCKI J.Catalytic ozonation in water:controversies and questions[J].Applied CatalysisB:Environmental,2013,142-143:465- 471.
[14]IKHLAQ A,BROWN D R,KASPRZYK H B.Mechanisms of catalytic ozonation on alumina and zeolites in water:formation of hydroxyl radicals[J].Applied Catalysis B:Environmental,2012,123-124:94- 106.[15]CARLOS A,GUZMAN P,JARED R.Kinetics of catalytic ozonation of atrazine in the presence of activated carbon[J].Separation and Purification Technology,2011,79(1):8- 14.
[16]LIU X Y,ZHOU Z M,JING G H.Catalytic ozonation of acid red B in aqueous solution over a Fe-Cu-Ocatalyst[J].Separation and Purification Technology,2013,15(1):129- 135.
[17]ZHANG T,CROUE J P.Catalytic ozonation not relying on hydroxyl radical oxidation:A selective and competitive reaction process related to metalcarboxylate complex[J].Applied Catalysis B:Environmental,2013,144(1):831- 839.
[18]PIRGALIOGLU S,OZBELGE T A.Comparison of non-catalytic and catalytic fast pyrolysis of corncob in a fluidized bed reactor [J].Bioresource Technology,2008,100(3):1428- 1434.
[19]AMUTHA R,SILLANPAA M,LEE G J.Catalytic ozonation of 2-ethoxy ethyl acetate using mesoporous nickel oxalates[J].Catalysis Communications,2013,43:88- 92.
[20]代欣欣,李汴生.水中臭氧溶解特性的研究[J].食品科技,2008,08:84- 87.DAI Xin-xin,LI Bian-sheng.Studies on solubility characteristics of ozone in water[J].Food Science and Technology,2008,08:84- 87.
[21]孟庆锐,安路阳,李超,等.O3催化氧化深度处理兰炭废水的试验研究[J].环境科学与技术,2013,02:133- 136.MENG Qing-rui,AN Lu-yang,LI Chao,et al.Experimental study on advanced treatment of semi-coking[J].Environmental Science&Technology,2013,02:133- 136.
[22]石枫华,马军.臭氧化和臭氧催化氧化工艺的除污效能[J].中国给水排水,2004,20(03):1- 4.SHI Feng-hua,MA Jun.Organics removal efficiency of ozonation and ozone catalyticoxidation process[J].China Water and Wastewater,2004,20(03):1- 4.
猜你喜欢
今日农业(2022年4期)2022-11-16
中国资源综合利用(2022年9期)2022-10-13
现代矿业(2022年3期)2022-04-09
煤气与热力(2021年10期)2021-12-02
湖南电力(2021年1期)2021-04-13
科学大观园(2020年18期)2020-09-16
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
皮革制作与环保科技(2020年14期)2020-03-17
制造技术与机床(2019年9期)2019-09-10
少儿科学周刊·少年版(2015年1期)2015-07-07
