
福州市性病门诊人群生殖道沙眼衣原体的分子流行病学研究
2015-03-17 01:12萧剑雄王惠榕
中国人兽共患病学报 2015年10期
萧剑雄,王惠榕
福州市性病门诊人群生殖道沙眼衣原体的分子流行病学研究
萧剑雄,王惠榕
目的 探讨福州市性病门诊人群泌尿生殖道沙眼衣原体的基因型分布状况。方法 收集2013-2014年临床疑似生殖道沙眼衣原体感染者标本2 019份,用实时荧光定量PCR检测沙眼衣原体,阳性标本用套式PCR扩增Omp1区并进行测序分析。测序结果上传至BLAST网站查找序列的相似性、构建系统树确定基因型。结果 检测沙眼衣原体阳性标本86份,成功分型的有83份,1株为混合感染。测得8个基因型,分别为:F型(36.59%)、E型(24.39%)、D型(12.20%)、G型(9.76%)、J型(9.76%)、H型(3.66%)、K型(2.44%)、B型(1.22%)。计算不同基因型间Omp1基因的核苷酸的同义突变率(dS)和非同义突变率(dN),发现仅G型的dN/dS值大于1,其他型均小于1。结论 F型和E型是福州市性病门诊人群泌尿生殖道沙眼衣原体的主要基因型。同义替代是Omp1基因在进化过程中的主要变异。
生殖道沙眼衣原体;基因分型;分子流行病学
据世界卫生组织估计,2008年全球成人泌尿生殖道沙眼衣原体新发病例达1.057亿。在发达国家生殖道沙眼衣原体感染的发病率已居于性传播疾病的首位[1]。衣原体感染的发病率在我国有逐年增加的趋势。通常衣原体感染的临床症状较轻,病情隐匿,但迁延难愈,相当一部分泌尿生殖道衣原体感染患者治疗效果不好,容易反复发作,最终可引起男性尿道炎、前列腺炎、附睾炎、直肠炎甚至引起不育;女性尿道炎、阴道炎、子宫颈炎、子宫内膜炎、输卵管炎和盆腔炎,甚至导致输卵管性不孕、异位妊娠等严重并发症,严重影响人们的健康和生活质量。衣原体的生殖道感染还可显著增加HIV感染的危险性[2],给社会造成了严重的经济负担己成为危害公共健康的一大问题。
感染人的沙眼衣原体包括两个生物学变种,一种为沙眼生物变种,感染柱状上皮细胞。另一种为性病淋巴肉芽肿变种,主要感染淋巴组织。各变种根据主要外膜蛋白(MOMP)抗原性不同,利用针对MOMP的单克隆抗体,将沙眼衣原体分成19种血清型。包括:4个淋巴肉芽肿血清型(L1,L2,L2a,和L3),15个沙眼生物型血清型(A到K,以及Ba、Da、Ia和Ja),其中A、B、Ba和C主要引起沙眼,而D到K则主要引起生殖道疾病,L1到L3引起性病性淋巴肉芽肿。目前,基因分型法已取代血清学分类法。在我国已有学者对广州、上海、深圳、南京等地的沙眼衣原体的基因分布进行了研究[3-6],但福州地区的分布情况还未见报道。本研究的目的是了解福州地区性病门诊人群生殖道沙眼衣原体的感染情况和基因分布,获得沙眼衣原体流行病学的相关信息,为疾病的预防与控制提供背景资料。
1 材料与方法
1.1 样本来源 2013年8月至2014年10月在福州市皮肤病防治院性病门诊就诊,临床疑似生殖道沙眼衣原体感染患者。诊疗时记录感染后有无宫颈或尿道异常分泌物、尿道刺痛、痒或灼热感等症状体征。
1.2 标本采集及保存 男性患者以男性采样拭子插入尿道约3~4 cm,旋转停留片刻后取出,女性患者先用无菌棉拭子擦去宫颈口外溢分泌物,然后用女性采样拭子插入宫颈管内约1~2 cm,滚动并停留30 s 取出。用中山大学达安基因股份有限公司的沙眼衣原体(CT)核酸检测试剂盒(PCR-荧光探针法)检测样本,将阳性样本冻存于-70 ℃冰箱备用。
1.3 主要试剂与仪器设备 细菌基因组DNA提取试剂盒(QIAamp DNA mini kit);Premix EX TaqTM Version 2.0 DNA 聚合酶(附加反应用缓冲液、dNTP混合液)购自宝生物工程大连有限公司;PCR引物由上海生工生物工程有限公司合成;其他试剂为国产分析纯。Biometra Tgradient PCR扩增仪为Biometra公司产品;水平电泳仪及电泳槽为北京六一仪器厂产品;凝胶成像仪为UVItec Limited公司产品。
1.4 方法
1.4.1 DNA提取 自-70℃冰箱中取出冻存的样本,将样本漂洗于1mL生理盐水,12 000 r/min离心10 min,细胞沉淀用QIAamp DNA mini kit 试剂盒提取基因组DNA,保存于-20 ℃冰箱中。
1.4.2 PCR扩增 参照文献设计[7]合成内外两对引物,外引物序列上游为P1:5′-ATG AAA AAA CTC TTG-3′,下游为OMP2:5′-ACT GTA ACT GCG TAT TTG TCT G-3′;内引物序列上游为MOMP87:5′-TGA ACC AAG CCT TAT GAT CGA CGG A-3′,下游为RVS1059:5′-GCA ATA CCG CAA GAT TTT CTA GAT TTC ATC-3′。扩增长度约为990 bp。第一轮PCR反应体系如下:PCR Premix 25 μL,引物P1和OMP2各5 μL(10 pmoL/L),模板DNA 5 μL,反应体系加灭菌蒸馏水至50 μL。反应条件:95 ℃预变性15 min,然后94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸90 s,共40个循环,最后末次延伸72 ℃ 7 min。取5 μL此PCR产物作模板,用内引物扩增, 第二轮扩增条将退火温度调整为60 ℃,余条件相同。两轮反应均设置水为阴性对照。PCR产物检测用1×TAE制备1.5%琼脂糖凝胶,其中含有1.0 μg/mL EB,将水平电泳仪电压调整为100 V,电泳时间为30 min。
1.4.3 测序 PCR产物送上海生工生物工程技术服务有限公司纯化后双向测序。测序引物为内引物。
1.4.4 进化树分析 应用Seqman 软件对已测序基因进行拼接,采用Blastn和Blastx程序,将测得的CT Omp1基因序列与美国国立医学图书馆NCBI主页(www.ncbi.nlm.nih.gov/blast)中的核苷酸序列及氨基酸序列进行同源性检索,并与标准CT参考株A/Sa1(M58938)、B/IU-1226(AF063208)、Ba/Apache 2(AF063194)、C/TW3(M17343)、D/B-120(X62918)、Da/TW-488(X62921)、E/Bour(X52557)、F/ICCal3(X52080)、G/UW57(AF063199)、H/Wash(X16007)、I/UW-12(AF063200)、Ia/IU-4168(AF063201、J/UW36(AF063202)、Ja/IU-A795(AF063203)、K/UW31(AF063204)、L1/440(M36533)、L2/434(M14738)、L3/404(X55700)进行同源性分析。用Mega5软件做进化树分析,以沙眼衣原体小鼠生物型MoPnT株(M64171)做外群。
2 结 果
2.1 检测阳性率 2013年8月至2014年10月共收集临床疑似生殖道沙眼衣原体感染者的标本2 019份,实时荧光定量PCR检测沙眼衣原体阳性标本86份,阳性率为4.26%。患者平均年龄为32.81±1.05岁。
2.2 Omp1基因分型结果 对实时荧光定量PCR阳性标本,扩增Omp1基因片段,有83份样本得到扩增,采用PCR产物直接测定法测定CT Omp1基因的核苷酸序列,将序列进行BLAST分析,根据其与各血清型参考株的同源性分型,共检出8个基因型,分布如下:F型30株(36.59%)、E型20株(24.39%)、D型10株(12.20%)、G型8株(9.76%)、J型8株(9.76%)、H型3株(3.66%)、K型2株(2.44%)、B型1株(1.22%),1株为混合感染不能确定其基因型。
2.3 Omp1基因进化树分析 同Millman K的研究结果[8],本研究临床菌株和参考株的Omp1基因的进化树也分为3个分支,即3个群:B群包括B、Ba、D、Da、E、L1、L2、L2a型;C群包括A、C、H、I、Ia、J、Ja、K、L3型;中间群包括F和G型。临床株未见A、Ba、C、Da、I、Ia、Ja、L1、L2、L2a、L3型。见图1。
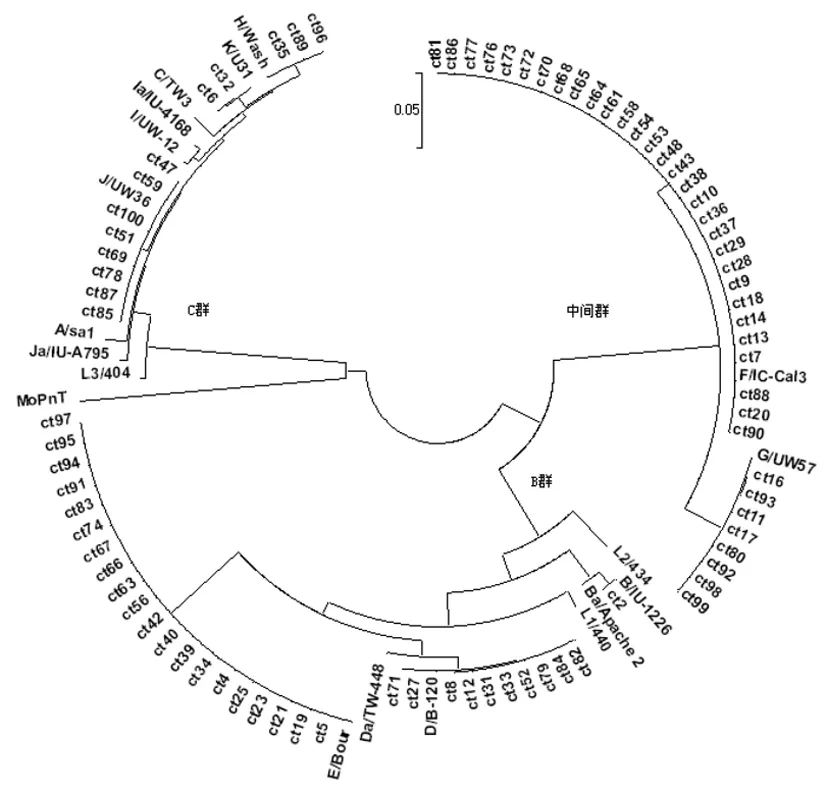
图1 基于生殖道沙眼衣原体Omp1基因构建的进化树
Fig.1 Phylogenetic reconstructions for the Omp1 gene ofC.trachomatis
2.4 Omp1基因序列进化距离分析 应用Mega5软件使用默认参数,对前述获得的Omp1基因的核酸序列按型别进行遗传距离分析,再分别计算dN和dS值以及dN/dS值,并据此分析该基因可能面对的环境选择压力状况。由于B、H、K型别的菌株较少而不予计算。分析可见G型、D型、J型的平均进化距离要大于F型和E型。其中G型的遗传距离最大,而E型最小。见图2。非同义替换的平均频率dN范围从0.000~0.482,同义替换的平均频率dS范围从0.000~0.625。仅有G型的dN/dS值大于1,其他型均小于1。
2.5 生殖道沙眼衣原体相关临床症状的基因型分布 感染后的症状体征分为有宫颈或尿道异常分泌物、尿道刺痛、痒或灼热感等相关临床症状和无症状两组,经统计分析,患者是否有相关临床表现在B群、C群及中间群间未见显著差异(χ2=1.304,P>0.05)。见表1。50例男性中无症状感染者22例,占44.00%;32例女性中无症状感染者19例,占59.38%,女性无症状感染者所占比例虽高于男性,但并无统计学差异(χ2=1.845,P>0.05)。无论男性还是女性F型均为最常见的基因型,其次是E型。F型在男性中占34.00%,女性中占40.63%,E型在男性中占22.00%,女性中占28.13%。B群、C群及中间群在男女性患者中的分布几率均等(χ2=0.006,P>0.05)。
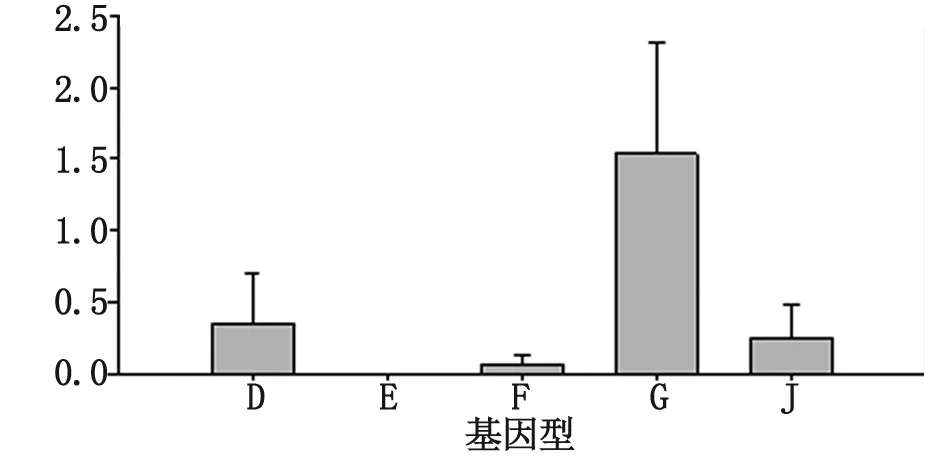
图2 生殖道沙眼衣原体基因型的平均进化距离
Fig.2 Mean distance within each genotypes ofC.trachomatis
3 讨 论
我们根据国内外同类型研究将生殖道沙眼衣原体基因型依据地区流行程度分为:E、F型为主要流行型,D、G、H、J和K型为次要基因型,其他B、Ba、I、Ia和Ja为少见基因型(见表2)。本研究发现福州生殖道沙眼衣原体同国内外其他地区报道的主要流行基因型一致以E、F型为主,表明沙眼衣原体的主要流行型别不存在地理分布的差异。但次要及少见基因型在不同国家或地区间的分布却不尽相同。G型在美国、瑞典、冰岛等国流行率较低[8,11-12],而在我国的流行率却较高。在美国Ia型取代I型流行[8],而在其他地区无论是I型还是Ia型均极少见。H型在台南和台北的地理分布存在差异[10]。据推测[13],可能由于E和F型自身的生物学优势增强其潜伏在宿主中的能力从而逃避宿主的免疫压力,也可能由于存在特异的粘附素或毒力因子使得它们在结构或功能上更有利于病原体的传播,而使得它们成为全球最流行的基因型。
学校以校院合作为平台,聘请行业、医院专家与学校专业教师组建专业建设指导委员会,参与专业开发,提出专业建设规划,确定重点专业和新专业的设置。2015年3月,学校与有关合作医院共同调研后决定新增农村医学、中医康复保健、中医护理、眼视光技术4个专业,现已获批准。
对Omp1基因核酸测序发现G、D和J型的平均进化距离要大于F和E型,说明是E、F型Omp1基因较稳定,变异度要远低于其他型。从分子生物学层面证实了上述推测即E、F型具有不同与其他型的生物学特性。我们的研究也表明除G型外,其他型别dN/dS值均小于1,在进化过程中大部分基因型的同义突变率要远远大于非同义突变率,说明未受到外界明显的选择压力的影响,Omp1基因突变为净化选择突变。
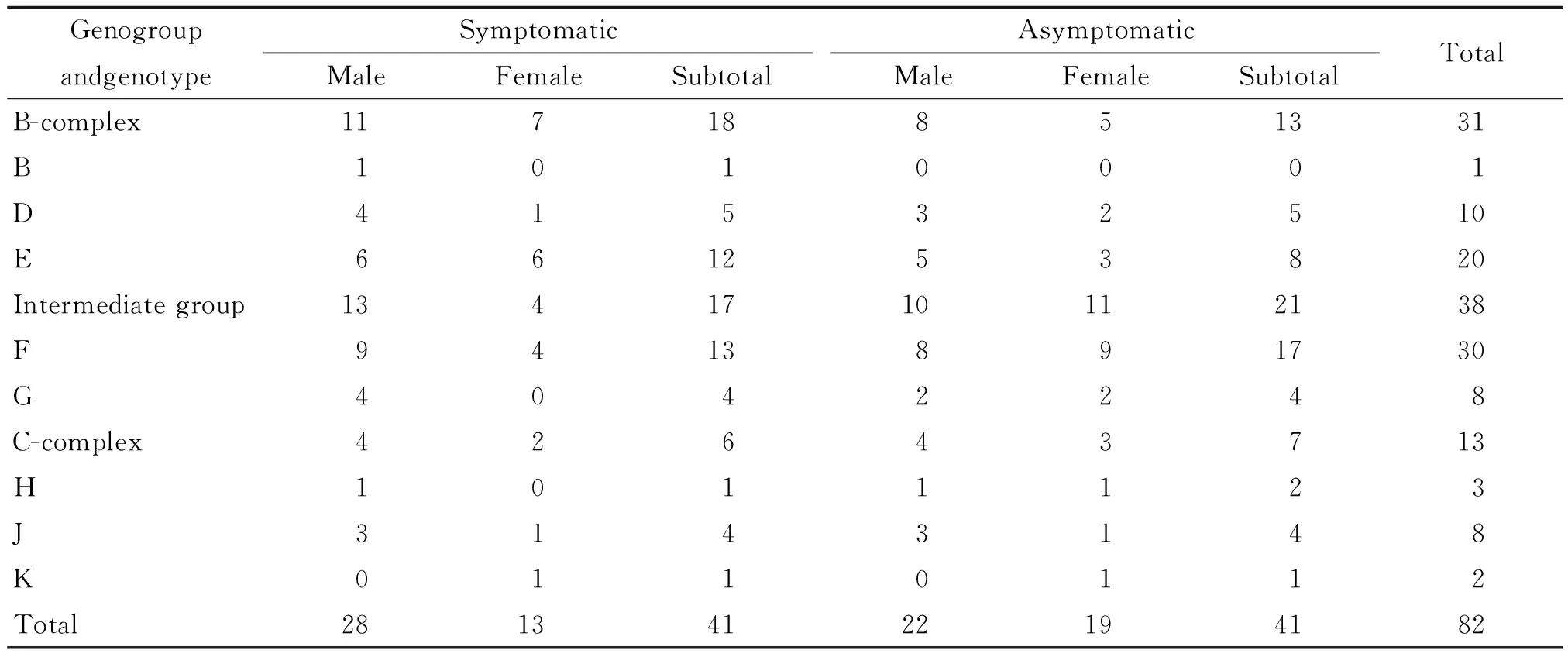
表1 生殖道沙眼衣原体相关临床症状的基因型分布
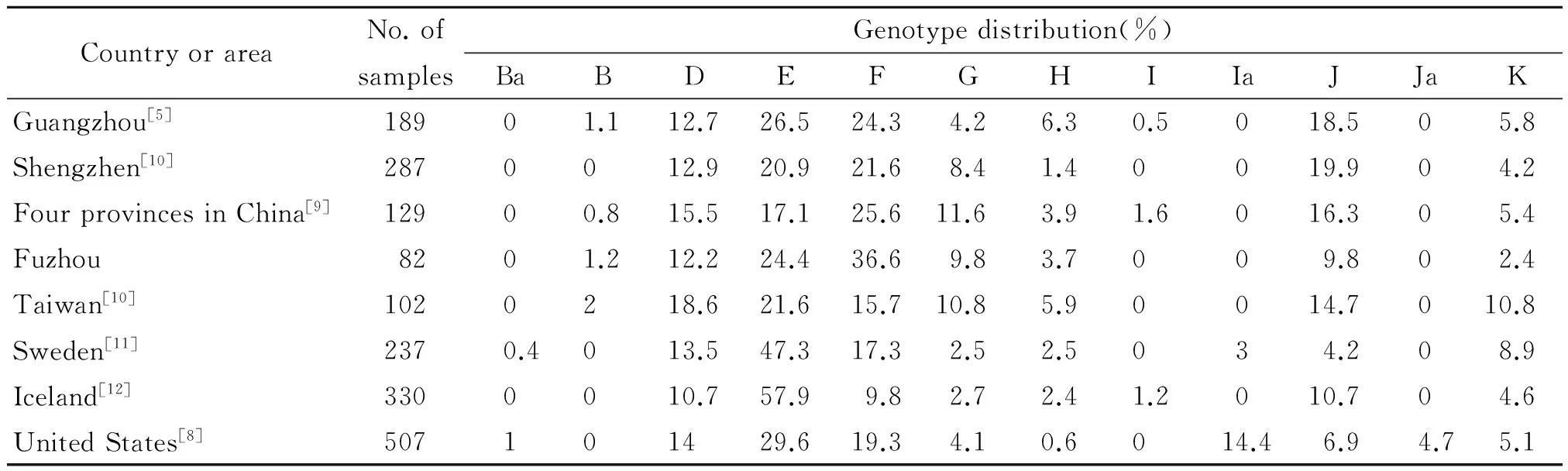
表2 国内外性病门诊者中生殖道沙眼衣原体基因型分布
由于E和F型大范围流行,有学者认为他们引起的免疫应答反应较弱,临床表现较轻,不易被检出而更容易扩散传播[14]。但Batteiger 等认为炎症表现与血清型无关[15],在我们的研究中,无论是男性或女性,没有发现基因型和临床体征存在联系,没有证据表明流行范围广的基因型,临床表现较轻。
我们注意到研究过程中存在的局限性。首先,由于商业试剂盒检测的是多拷贝质粒,而用于基因分型的Omp1基因是单拷贝基因,扩增的敏感性较低。因此,有可能实时荧光PCR检测阳性的标本,而在扩增Omp1基因时有可能得到阴性结果。其次,用PCR产物直接测序的局限性在于不能识别混合基因型的序列图谱,除非对PCR产物克隆测序。第三,基因型的分布可能会受到目标人群样本数的影响。
总之,E、F型为福州地区生殖道沙眼衣原体的优势基因型。这一发现可以完善福州地区生殖道沙眼衣原体监测的基线数据,但仍需进一步扩大样本量。
[1]World Health Organization. Global incidence and prevalence of selected curable sexually transmitted infections-2008[EB/OL]. http://www.who.int/topics/sexually_transmitted_infections/, 2015-04-12/2015-05-21.
[2]Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice:the contribution of other sexually transmitted diseases to sexual transmission of HIV infection[J]. Sex Transm Infect, 1999, 75(1):3-17.
[3]Gao X, Chen XS, Yin YP, et al. Distribution study ofChlamydiatrachomatisserovars among high-risk women in China performed using PCR-restriction fragment length polymorphism genotyping[J]. J Clin Microbiol, 2007, 45(4):1185-1189.
[5]Yang B, Zheng HP, Feng ZQ, et al. The prevalence and distribution ofChlamydiatrachomatisgenotypes among sexually transmitted disease clinic. Patients in Guangzhou, China, 2005-2008[J]. Jpn J Infect Dis, 2010, 63:342-345.
[6]Zhang JJ, Zhao GL, Wang F, et al. Molecular epidemiology of genitalChlamydiatrachomatisinfection in Shenzhen, China[J]. Sex Transm Infect, 2012, 88(4):272-277.
[7]Lysen M, Osterlund A, Rubin CJ, et al. Characterization of ompA genotypes by sequence analysis of DNA from all detected cases ofChlamydiatrachomatisinfections during 1 year of contact tracing in a Swedish county[J]. J Clin Microbiol, 2004, 42(4):1641-1647.
[8]Millman K, Black CM, Johnson RE, et al. Population-based genetic and evolutionary analysis ofChlamydiatrachomatisurogenital strain variation in the United States[J]. J Bacteriol, 2004, 186(8):2457-2465.
[9]Han Y, Yin YP, Shi MQ, et al. Difference in distribution ofChlamydiatrachomatisgenotypes among different provinces a pilot study from four provinces in China[J]. Jpn J Infect Dis, 2013, 66(1):69-71.
[10]Hsu MC, Tsai PY, Chen KT, et al. Genotyping ofChlamydiatrachomatisfrom clinical specimens in Taiwan[J]. J Med Microbiol, 2006, 55(Pt 3):301-308.
[11]Characterization ofChlamydiatrachomatisomp1 genotypes among sexually transmitted disease patients in Sweden[J]. J Clin Microbiol, 2001, 39(11):3915-3919.
[12]Jonsdottir K, Kristjansson M, Hjaltalin Olafsson J, et al. The molecular epidemiology of genitalChlamydiatrachomatisin the Greater Reykjavik Area, Iceland[J]. Sex Transm Dis, 2003, 30(3):249-256.
[13]Nunes A, Borrego MJ, Nunes B, et al. Evolutionary dynamics of ompA, the gene encoding theChlamydiatrachomatiskey antigen[J]. J Bacteriol, 2009, 191(23):7182-7192.
[14]Stothard DR, Boguslawski G, Jones RB. Phylogenetic analysis of theChlamydiatrachomatismajor outer membrane protein and examination of potential pathogenic determinants[J]. Infect Immun, 1998, 66(8):3618-3625.
[15]Batteiger BE, Lennington W, Newhall WJ, et al. Correlation of infecting serovar and local inflammation in genital chlamydial infections[J]. J Infect Dis, 1989, 160:332-336.
Molecular epidemiology ofChlamydiatrachomatisin outpatients visiting STD clinics in Fuzhou, China
XIAO Jian-xiong,WANG Hui-rong
(FujianCenterforDiseaseControlandPrevention,Fuzhou350001,China)
We investigated the distribution ofChlamydiatrachomatisgenotypes in the outpatients visiting STD clinics in Fuzhou City, China. Between 2013 and 2014, a total of 2 019 urethral or endocervical specimens from susceptible genitalC.trachomatisinfections were collected and detected by using real-time PCR assay kit. TheC.trachomatis-positive specimens were subsequently amplified with nested PCR for Omp1 region and analyzed by sequencing. Genotyping was performed by BLAST similarity search and phylogenetic tree analysis. Eighty-three of the 86C.trachomatis-positive specimens were successfully sequenced. Only one specimen was identified as multiple infection. Eight different genotypes were identified. The most prevalent was F (36.59%), followed by E (24.39%), D (12.20%), G (9.76%), J (9.76%), H (3.66%), K (2.44%) and B (1.22%). Synonymous mutation rate and non-synonymous mutation rate among Omp1 genes of different genotypes were calculated. All genotypes but G showed dN/dSvalues of<1. Genotypes F and E were commonest. Synonymous substitution was the major pattern of variation in the process of Omp1 gene evolution.
Chlamydiatrachomatis; genotyping; molecular epidemiology
Wang Hui-rong, Email:fjwhr@hotmail.com
王惠榕,EmaiL:fjwhr@hotmail.com
福建省疾病预防控制中心,福州 350001
10.3969/j.issn.1002-2694.2015.10.011
R374
A
1002-2694(2015)10-0947-05
2015-04-14;
2015-06-23

猜你喜欢
动物医学进展(2022年11期)2022-12-28
中华养生保健(2020年2期)2020-11-16
家庭科学·新健康(2020年6期)2020-07-06
中国生殖健康(2019年2期)2019-08-23
中老年保健(2019年9期)2019-01-13
妈妈宝宝(2018年4期)2018-11-29
现代养生·下半月(2018年5期)2018-09-10
中南医学科学杂志(2018年3期)2018-01-17
妇女之友(2016年5期)2016-05-14
家庭医药·快乐养生(2015年8期)2015-09-10
