
鄱阳湖湿地不同土地利用方式下土壤微生物群落功能多样性
2015-03-10 12:22刘以珍
生态学报 2015年4期
张 杰,胡 维,刘以珍,葛 刚,2,吴 兰,2,*
1 江西省分子生物学与基因工程重点实验室,南昌大学, 南昌 330031 2 鄱阳湖环境与资源利用教育部重点实验室,南昌大学, 南昌 330031
鄱阳湖湿地不同土地利用方式下土壤微生物群落功能多样性
张 杰1,胡 维1,刘以珍1,葛 刚1,2,吴 兰1,2,*
1 江西省分子生物学与基因工程重点实验室,南昌大学, 南昌 330031 2 鄱阳湖环境与资源利用教育部重点实验室,南昌大学, 南昌 330031
于2011年5月分别采集鄱阳湖围垦92、48a和38a的水稻田,退田还湖25a的退耕地以及自然湿地共5个样地的表层土壤,利用Biolog-ECO板技术对土壤微生物群落的单一碳源利用情况进行了测定,并结合群落指数和主成分分析(PCA)对培养72 h土壤微生物群落功能多样性变化进行了分析。结果表明:退耕地和自然湿地土壤微生物群落利用31种碳源的能力较强,来自不同围垦年限的土壤微生物群落利用碳源能力均较弱;且随围垦时间的增长,土壤微生物对碳源的利用能力呈降低的趋势。自然湿地、退耕地与围垦92、38a样地土壤之间存在显著的微生物功能多样性差异;围垦对土壤微生物代谢糖类、羧酸类、氨基酸类物质的影响最为明显。结果提示,围垦改变了湿地土壤微生物群落结构,退田还湖有助于湿地土壤微生物群落结构的恢复。
鄱阳湖;湿地;围垦年限;退田还湖;Biolog
湿地是一种常年或季节性积水或过湿的独特生态系统。它具有一系列重要的生态功能,包括为野生动植物提供栖息地,贮存来自降雨的水源[1],防止碳氮养分释放到邻近地表水而造成富营养化[2],净化市政工农业排放的废水。因而湿地被称为“地球之肾”。而湿地生态系统对污染物的转化作用主要与其土壤中种类繁多的好氧和厌氧微生物有关[3]。因此对于湿地土壤微生物的研究具有重大意义。
围垦是对湿地资源开发利用的主要形式之一,可以缓减土地资源的短缺。但围垦等人类活动改变了土壤环境,对湿地生态系统造成干扰,进而对湿地的微生物群落产生影响[4]。那么围垦与否对湿地土壤微生物群落结构究竟有何影响?不同围垦年限下湿地微生物的群落变化又有何规律?退田还湖是否能有效修复湿地?目前国内外鲜见对该方面的研究。
本研究选取鄱阳湖典型自然湿地、不同围垦年限水稻田以及退耕地为研究对象,利用Biolog-ECO板技术对其土壤微生物群落功能多样性进行了研究,旨在初步揭示土壤微生物群落功能多样性随围垦时间的变化规律,并从土壤微生物层面对退田还湖这一湿地修复措施的可行性提供一定的理论依据。
1 材料与方法
1.1 样地概况与样品采集
样地位于鄱阳湖西南角的南昌县蒋巷联圩和南矶山湿地国家自然保护区(28°43′40″—29 °00′17″N,115°55′36″—116°22′19″E),属赣江尾闾入湖三角洲。区内年平均降雨量为1470 mm,年平均气温为16—19 ℃,光照充足,属于亚热带季风气候。
于2011年5月进行样品采集,沿围垦时间序列依次选取围垦92a水稻田(RL92)、围垦48a水稻田(RL48)、围垦38a水稻田(RL38)、退田还湖25a的退耕地(RC25)和南矶山自然湿地(NW)5个梯度样地。RC25样地的覆盖植被为苔草群落(Carexcinerascens)。NW样地的主要植被为苔草和虉草群落(Phalarisarundinacea)。每个样地设立3个平行样点,样点之间的距离不小于1 km,每个样点中进行3次重复取样,3次重复之间的距离不小于100 m,现场将3次重复土样混匀,共得到15个土壤样品。采样工具为土钻,取样前先将土壤表面的植被小心去除,取样深度0—20 cm,土样装入无菌硬质塑料盒,并用冰盒迅速运回实验室4 ℃保存。
1.2 土壤微生物功能多样性分析
参照文献[5]的方法测定各样地土壤的理化参数(表1)。采用Biolog-ECO微平板法对不同土地利用方式下土壤的微生物功能多样性进行分析。具体实验步骤参照Govaerts等的研究方案[6]。ECO板放置于生化培养箱中25 ℃恒温培养10 d,每隔12 h在Biolog微生物自动鉴定系统(MicroStationTMSystem,美国Biolog公司)上测定590 nm处的吸光值。
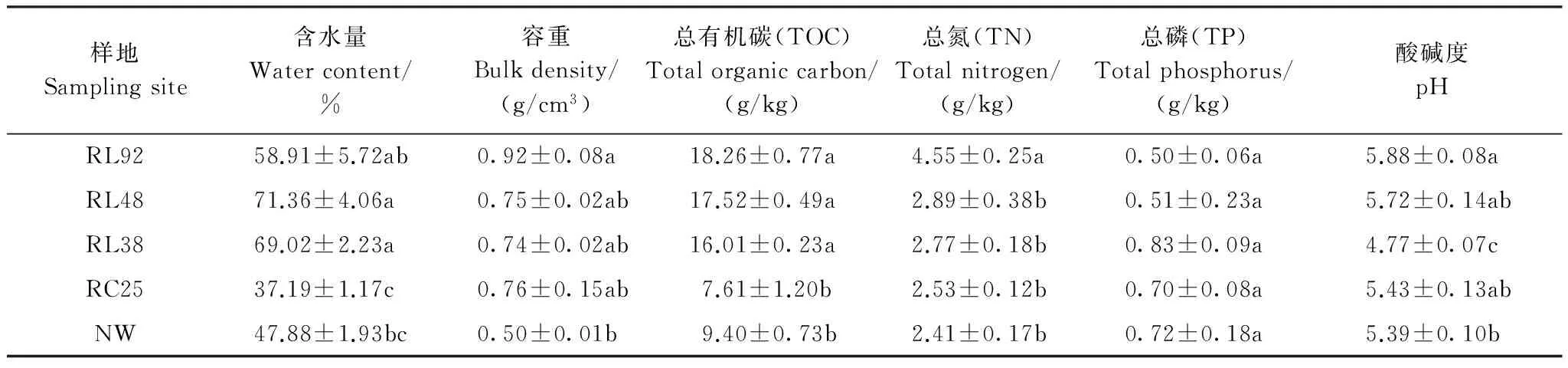
表1 不同土地利用方式下土壤理化参数Table 1 Physicochemical parameters of soil across land-use types
RL92:围垦92a水稻田;RL48:围垦48a水稻田;RL38:围垦38a水稻田;RC25:退田还湖25a的退耕地;NW:南矶山自然湿地
1.3 数据分析
将每次读板得到的吸光值都减去培养0 h时的吸光值,以去除本底干扰[7],负值全部归0。然后分别计算平均颜色变化率(AWCD)和培养72 h(对数期)后的Shannon-Weaver多样性指数(H)[8],并用OD590>0.15的微孔数作为相应样品土壤细菌群落代谢丰富度(S)[6]。运用SPSS 19.0对数据进行差异性分析和主成分分析(PCA)。
2 结果与分析
2.1 不同土地利用方式下土壤微生物群落AWCD
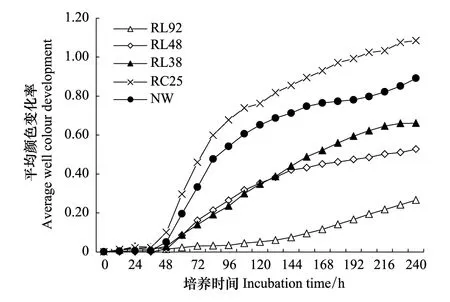
图1 不同土地利用方式下土壤微生物群落AWCD随培养时间的变化Fig.1 Variation of microbial AWCD against incubation time for soil across land-use typesRL92:围垦92a水稻田;RL48:围垦48a水稻田;RL38:围垦38a水稻田;RC25:退田还湖25a的退耕地;NW:南矶山自然湿地
平均颜色变化率(AWCD)反映了微生物群落对Biolog-ECO板中单一碳源的整体利用能力,是微生物群落功能多样性的一个重要指标[9]。如图1所示,不同土地利用方式下土壤微生物群落AWCD的变化趋势均为随培养时间的增加而升高。在0—48 h内各样地土壤的AWCD值均较低,48 h以后,除RL92升幅平缓以外,其他样地的AWCD都迅速升高。说明土壤微生物对碳源利用开始于48 h以后。总体上,不同土地利用方式下土壤微生物利用31种碳源的AWCD值呈RC25 > NW > RL38 > RL48 > RL92的变化趋势,值得注意的是,RC25与NW的AWCD值无显著差异(P>0.05),但两者与其他围垦样地均有显著差异(P<0.01)。
2.2 不同土地利用方式下土壤微生物群落碳源利用特征
Biolog-ECO板中含有多聚物(4种)、糖类(10种)、羧酸类(7种)、酚酸类(2种)、氨基酸类(6种)和胺类(2种)共6大类31种单一碳源。通过分析每一类碳源的AWCD值可以判断各样地土壤微生物对特定类别碳源的利用能力[10]。不同土地利用方式下土壤微生物利用6类碳源的AWCD随培养时间的变化如图2。图中显示,不同样地土壤微生物利用6类碳源的AWCD都随培养时间的延长而大致呈现升高趋势,且土壤微生物代谢6类碳源的能力均具有显著差异(P<0.001)。
对于多聚物类碳源的利用(图2),来自RC25的微生物AWCD最高,NW和RL48居中,RL38和RL92最低。对于糖类、羧酸类和氨基酸类碳源,土壤微生物AWCD变化规律与上述31种碳源利用总体情况类似,均表现为RC25 > NW > RL38 > RL48 > RL92,其中围垦样地土壤微生物利用糖类、氨基酸类碳源的AWCD显著低于RC25和NW样地,且随着围垦年限的增加,土壤微生物群落代谢糖类、氨基酸类物质的能力逐渐降低,而RC25和NW样地的微生物代谢能力无显著差异;对于羧酸的利用,值得注意的是RL92的AWCD极小,显著低于RL38和RL48,说明围垦达到一定年限后,土壤微生物对羧酸类物质的代谢能力急剧下降。对于酚酸类碳源,RC25、NW和RL38显著高于RL48和RL92,说明土壤微生物利用酚酸类碳源能力的改变发生于围垦38a至48a间。土壤微生物利用胺类碳源的能力具有显著特征,除RL92的AWCD显著小于其他样地外,其他样地间微生物AWCD均无显著差异,说明不同土地利用方式下土壤微生物降解胺类物质的能力较稳定,该物质降解能力改变的时间阈值可能是围垦92a(图2)。

图2 不同土地利用方式下土壤微生物利用各类碳源的AWCD变化Fig.2 Variation of microbial carbon utilization AWCD for soil across land-use types
2.3 不同土地利用方式下土壤微生物群落功能多样性指数和丰富度
Shannon-Weaver多样性指数(H)和丰富度指数(S)可以反映土壤微生物群落的功能多样性[11]。表2为不同土地利用方式下土壤微生物群落Biolog-ECO板实验培养72 h后的代谢多样性指数和丰富度指数。由表可知,各样地土壤微生物群落的香农多样性指数无显著差异,但丰富度指数在样地间存在显著差异(P<0.01)。丰富度S的最小值出现在RL92,其次是RL38,RC25和NW的丰富度最高;但值得注意的是,RL48的丰富度也较高,且与RC25和NW无显著差异。总体而言,围垦水稻田土壤微生物群落丰富度均小于自然湿地,而退耕地的土壤微生物群落丰富度则大于自然湿地。

表2 不同土地利用方式下土壤微生物群落指数(n=3)Table 2 Microbial community indices of soil across land-use types
2.4 不同土地利用方式下土壤微生物群落代谢主成分分析
对不同土地利用方式下土壤微生物72 h利用碳源的光密度值进行PCA分析。按照提取的主成分一般要求累计方差贡献率达到85%[12]并且其特征根大于1[13]这两个原则,共提取了8个主成分(PC),累积方差贡献率为91.03%。其中第1主成分(PC1)和第2主成分(PC2)的方差贡献率分别为31.28%和21.94%。第3至第8主成分的方差贡献率相对较小,分别为10.15%、6.90%、6.70%、5.92%、4.19%和3.96%。
不同土地利用方式下土壤微生物碳源利用的PCA排序图如图3。由图可知,前两个主成分共解释了53.22%的变异。结合方差分析结果表明,各样地在PC1上没有明显的空间分异。而在PC2上,5个样地的土壤微生物群落明显分为2簇:RL92和RL38聚为1簇,NW和RC25聚为另一簇,RL48居于2簇之间。PCA分析显示,来自RL92和RL38样地与来自NW和RC25样地的土壤微生物群落结构存在显著差异,从图中还可以直观地看出,在PC2上,各样地土壤微生物碳源利用的主成分分析得分排序为:RC25 > NW > RL48 > RL38 > RL92,这与表2的群落丰富度指数分析结果相一致。
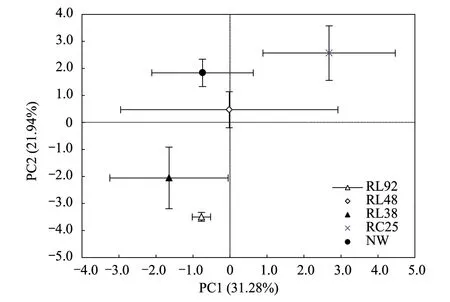
图3 不同土地利用方式下土壤微生物群落功能主成分分析Fig.3 Principal component analysis for microbial community function of soil across land-use types
对所提取的8个主成分,分别计算6类碳源的主成分得分F和样地的综合得分zF,结果如图4。其中F值的相对大小可以表征不同样地土壤微生物群落对同一类碳源的代谢能力和同一样地土壤微生物对不同类型碳源代谢能力的高低,而zF的相对大小则可以反映不同样地土壤微生物对Biolog-ECO板31种碳源综合代谢能力的高低。
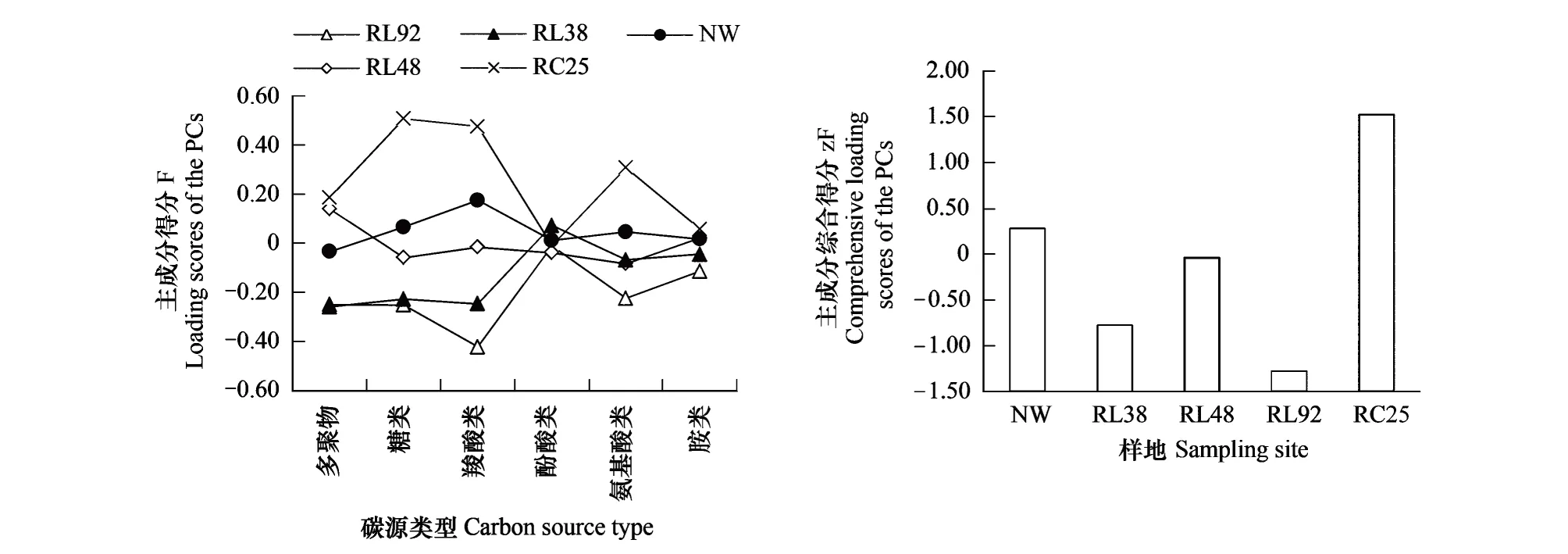
图4 各样地碳源类型主成分得分以及综合得分Fig.4 Loading scores and comprehensive loading scores of the first eight principal components
对图4中的F值进行横向比较发现:不同土地利用方式下土壤微生物利用碳源的能力不尽相同。对于RC25,其代谢糖类、羧酸类和氨基酸类碳源的能力较强,代谢酚酸类的能力较弱;对于NW,其对羧酸类的代谢能力最强,而对多聚物的利用能力最弱;对于RL38,其代谢酚酸类的能力最强,而代谢多聚物、糖类、羧酸类碳源的能力最弱;对于RL48,其利用多聚物类碳源的能力最强,而利用糖类和氨基酸类碳源的能力最弱;对于RL92,其土壤微生物群落对酚酸类碳源的代谢能力最强,而对羧酸类碳源的代谢能力最弱;对F值进行纵向比较可知:代谢多聚物、糖类、羧酸类、氨基酸类和胺类能力最强的都是来自RC25的土壤微生物,代谢以上物质能力最弱的是来自RL92的土壤微生物;代谢酚酸类物质能力最强的是RL38,最弱的是RL48。且围垦水稻田在糖类、羧酸类和氨基酸类的F值明显小于自然湿地和退耕地。
从图4样地主成分综合得分zF可以看出,5个样地土壤微生物对Biolog-ECO板中31种单一碳源的综合代谢能力排序为:RC25 > NW > RL48 > RL38 > RL92。即退耕地>自然湿地>围垦水稻田。
3 讨论与结论
土壤微生物群落利用31种碳源的能力随着围垦年限的增长(依次为自然湿地、围垦38a、围垦48a和围垦92a)整体呈下降趋势(自然湿地可看作围垦0a),这是因为围垦耕作可能会造成土壤团聚体的分裂和表层土壤中有机质的损耗,从而导致土壤微生物代谢多样性的下降[14]。此外,围垦耕作后种植的作物品种单一(水稻),使得围垦土壤与自然湿地土壤的植物群落结构显著不同,而植物种类会影响土壤中的碳分配和微生物多样性[15]。值得注意的是,基于培养72 h(对数期)数据得到的微生物代谢丰富度指数和PCA分析结果都表明,围垦48a的水稻田土壤微生物群落对碳源的利用能力介于自然湿地和围垦38a之间,其代谢活性、丰富度指数均高于围垦38a的水稻田,且与自然湿地的相近。可能的原因有:
1)围垦过程伴随着土壤微生物群落的演替,即由典型的湿地土壤微生物群落逐渐演替为典型的耕作土壤微生物群落,Schipper等发现微生物代谢多样性跟碳输入有关,随着演替过程中碳输入的增加,土壤微生物代谢多样性呈现演替初期较低、之后升高、最后又下降的趋势。根据“中度干扰假说”来解释,演替初期过低的碳输入抵制土壤微生物多样性,过高的碳输入则加剧微生物竞争,从而降低其多样性[16]。结合表1中总有机碳(TOC)含量可推断,本研究不同围垦时间序列下土壤微生物的演替也可能存在类似情况;
2)作为研究土壤微生物群落代谢功能的技术,Biolog有其局限性。ECO板中的31种碳源并不能全部反映自然条件下土壤微生物所处的复杂底物环境,所以Biolog技术不能够完整地反映土壤中微生物群落的功能多样性和复杂性[17]。
从土地利用类型的角度来看,退耕地土壤微生物群落利用碳源的能力最强,其丰富度指数和主成分综合得分zF值均为最大,自然湿地次之,围垦水稻田的最低。说明退田还湖在一定程度上有助于土壤微生物群落向着自然湿地的状态恢复。6类碳源的AWCD曲线和主成分得分F的分析结果都初步表明,围垦对土壤中与糖类、羧酸类和氨基酸类物质代谢有关的微生物影响最为明显,围垦年限越长,湿地土壤微生物对这3类碳源的利用能力越差。
从各样地土壤的理化参数(表1),可知鄱阳湖湿地不同土地利用方式下土壤理化参数除TP外,含水量、容重、TOC、TN(总氮)和pH值都存在显著差异,且总体上均为围垦水稻田大于退耕地和自然湿地。特别是TOC,围垦水稻田土壤TOC含量显著高于自然湿地和退耕地(P<0.001),且围垦年限越长,含量越高,而自然湿地和退耕地之间无显著差异,这与Iost等[18]的研究结果一致,即长期围垦有助于土壤有机碳的积累,而退田还湖后土壤有机碳损耗增加。其主要原因可能是农作物腐殖质和根系分泌物的不断积累,但微生物在此过程中所起的作用尚需进一步研究。此外,典范对应分析(CCA)和相关性分析(数据未列出)结果表明,各理化参数与土壤微生物代谢多样性之间并未显示出规律性或显著性关系。
本研究利用Biolog-ECO板技术对鄱阳湖湿地不同土地利用方式下土壤微生物群落的代谢多样性进行了研究,得到了一些初步的结果。今后的工作将利用454高通量测序结合生态学技术,围绕不同季节不同土地利用方式下土壤微生物群落组成和结构、功能微生物组成及其与理化参数的关系等方面展开。以期更为深入、全面地阐释围垦和退田还湖对湿地土壤微生物群落结构和组成的影响。
[1] Mitsch W J, Gosselink J G. Wetlands. New York: John Wiley & Sons, 2000.
[2] Vitousek P M, D′antonio C M, Loope L L, Westbrooks R. Biological invasions as global environmental change. American Scientist, 1996, 84(5): 468- 478.
[3] D′Angelo E M, Reddy K R. Regulators of heterotrophic microbial potentials in wetland soils. Soil Biology and Biochemistry, 1999, 31(6): 815- 830.
[4] Mentzer J L, Goodman R M, Balser T C. Microbial response over time to hydrologic and fertilization treatments in a simulated wet prairie. Plant and Soil, 2006, 284(1/2): 85- 100.
[5] 鲁如坤. 土壤农业化学分析方法. 北京: 中国农业科技出版社, 2000..
[6] Govaerts B, Mezzalama M, Unno Y, Sayre K D, Luna-Guido M, Vanherck K, Dendooven L, Deckers J. Influence of tillage, residue management, and crop rotation on soil microbial biomass and catabolic diversity. Applied Soil Ecology, 2007, 37(1/2): 18- 30.
[7] Garau G, Castaldi P, Santona L, Deiana P, Melis P. Influence of red mud, zeolite and lime on heavy metal immobilization, culturable heterotrophic microbial populations and enzyme activities in a contaminated soil. Geoderma, 2007, 142(1/2): 47- 57.
[8] Wei Y, Yu L F, Zhang J C, Yu Y C, Deangelis D L. Relationship between vegetation restoration and soil microbial characteristics in degraded karst regions: a case study. Pedosphere, 2011, 21(1): 132- 138.
[9] Wei D, Yang Q, Zhang J Z, Wang S, Chen X L, Zhang X L, Li W Q. Bacterial community structure and diversity in a black soil as affected by long-term fertilization. Pedosphere, 2008, 18(5): 582- 592.
[10] Preston-Mafham J, Boddy L, Randerson P F. Analysis of microbial community functional diversity using sole-carbon-source utilization profiles - a critique. FEMS Microbiology Ecology, 2002, 42(1): 1- 14.
[11] Feng S G, Zhang H X, Wang Y F, Bai Z H, Zhuang G Q. Analysis of fungal community structure in the soil of Zoige Alpine Wetland. Acta Ecologica Sinica, 2009, 29(5): 260- 266.
[12] 郝黎仁,樊元,郝哲欧. SPSS实用统计分析. 北京: 中国水利水电出版社, 2003.
[13] Zhang H F, Li G, Song X L, Yang D L, Li Y J, Qiao J, Zhang J N, Zhao S L. Changes in soil microbial functional diversity under different vegetation restoration patterns for Hulunbeier Sandy Land. Acta Ecologica Sinica, 2013, 33(1): 38- 44.
[14] Lupwayi N Z, Rice W A, Clayton G W. Soil microbial diversity and community structure under wheat as influenced by tillage and crop rotation. Soil Biology and Biochemistry, 1998, 30(13): 1733- 1741.
[15] Ladygina N, Hedlund K. Plant species influence microbial diversity and carbon allocation in the rhizosphere. Soil Biology and Biochemistry, 2010, 42(2): 162- 168.
[16] Schipper L A, Degens B P, Sparling G P, Duncan L C. Changes in microbial heterotrophic diversity along five plant successional sequences. Soil Biology and Biochemistry, 2001, 33(15): 2093- 2103.
[17] Kong W D, Zhu Y G, Fu B J, Marschner P, He J Z. The veterinary antibiotic oxytetracycline and Cu influence functional diversity of the soil microbial community. Environmental Pollution, 2006, 143(1): 129- 137.
[18] Iost S, Landgraf D, Makeschin F. Chemical soil properties of reclaimed marsh soil from Zhejiang Province PR China. Geoderma, 2007, 142(3- 4): 245- 250.
Response of soil microbial functional diversity to different land-use types in wetland of Poyang Lake, China
ZHANG Jie1, HU Wei1, LIU Yizhen1, GE Gang1,2, WU Lan1,2,*
1KeyLaboratoryofMolecularBiologyandGeneticEngineeringinJiangxiProvince,NanchangUniversity,Nanchang330031,China2KeyLaboratoryofEnvironmentandResourceUtilizationofPoyangLake,MinistryofEducation,Nanchang330031,China
Wetland ecosystem, the transitional region between terrestrial ecosystem and aquatic ecosystem, plays a crucial role in global carbon and nitrogen cycle, flood constraint, runoff regulation, climate improvement, pollution prevention and organismal habitats supply. However, as a consequence of human beings′ reclamation and contamination in the past centuries, wetland ecosystems around the world have been destructed and their total area has being declining. Hence, it is urgent to restore wetland ecosystem resources and its ecological function. Microorganisms are one of the predominant participants associated with the function of wetlands. To accomplish wetland restoration scientifically and efficiently, it is indispensable to reveal the responding mechanism of microbial variation due to wetlands′ reclamation and restoration. In this study, a series of lands under different utilized types were selected as sampling sites in Nanjishan Wetland National Nature Reserve of Poyang Lake, which located in Jiangxi Province, China. The sites were 38, 48 and 92 year-old reclaimed lands (named RL38, RL48 and RL92, all were paddy fields), 25-year-old retired cropland (RC25) and native wetland (NW). In May 2011, surface soil samples (0—20 cm depth) were collected using earth boring auger in these sampling sites. Biolog-ECO plates were performed to survey the sole-carbon-source utilization of soil microbial communities. Average well colour development (AWCD) of all 31 carbon sources and the 6 kinds of carbon sources were calculated respectively. Microbial community indices calculation and principal components analysis (PCA) were carried out to analyze the variations of functional diversity of soil microbial community in exponential phase. It aimed to preliminarily reveal: 1) the effects of reclamation on the functional diversity of soil microbial community; 2) whether it was efficient at microbial level for wetland restoration by returning farmland to lake wetland. In consequence, 1) the average well colour development (AWCD) of all the soil samples was at a low level during the initial 48 hours. Subsequently, all the AWCDs presented a rapid rising in addition to the 92-year reclaimed land. 2) Generally, the order of AWCD of soil microorganisms was as follows: RC25 > NW > RL38 > RL48 > RL92. Notably, it was significantly higher in retired cropland and native wetland than in reclaimed lands (P<0.01). 3) The AWCD variation of carbohydrates, carboxylic acids and amino acids among sampling sites accorded with the total AWCD of all the 31 sole-carbon sources. 4) According to the microbial functional Richness index and the comprehensive loading scores of PCA, the utilization ability order in exponential phase among sampling sites was RC25 > NW > RL48 > RL38 > RL92. On the first two axes, a total 53.22% variation of functional diversity was explained. The results indicated that 1) the functional diversity of soil microbial community was significantly lower in reclaimed lands than in native wetland and decreased with the extension of reclamation age; 2) soil microorganisms associated with metabolism of carbohydrates, carboxylic acids and amino acids were most apparently affected by reclamation. 3) it was efficient, to some extent, to recover the soil microbial metabolic activity by returning the farmland to lake wetland.
Poyang Lake; wetland; reclamation age; returning farmland to lake wetland; biolog
国家自然科学基金(31360127, 31260110)
2013- 06- 12;
日期:2014- 04- 11
10.5846/stxb201306121688
*通讯作者Corresponding author.E-mail: wl690902@hotmail.com
张杰,胡维,刘以珍,葛刚,吴兰.鄱阳湖湿地不同土地利用方式下土壤微生物群落功能多样性.生态学报,2015,35(4):965- 971.
Zhang J, Hu W, Liu Y Z, Ge G, Wu L.Response of soil microbial functional diversity to different land-use types in wetland of Poyang Lake, China.Acta Ecologica Sinica,2015,35(4):965- 971.
猜你喜欢
风流一代·青春(2022年7期)2022-07-22
环境工程技术学报(2022年3期)2022-06-05
云南化工(2021年10期)2021-12-21
昆钢科技(2021年6期)2021-03-09
建材发展导向(2021年24期)2021-02-12
西南农业学报(2016年5期)2016-05-17
电源技术(2016年9期)2016-02-27
中国塑料(2015年2期)2015-10-14
现代农业(2015年5期)2015-02-28
中国塑料(2014年1期)2014-10-17
