
微流控芯片非接触电导法测定僵蚕中草酸铵的含量Δ
2015-03-10 02:34杨秀娟陈缵光陈倩瑜南方医科大学珠江医院广州51080中山大学药学院广州510006佛山市顺德区北滘医院广东佛山5811
中国药房 2015年15期
杨秀娟,陈缵光,陈倩瑜(1.南方医科大学珠江医院,广州 51080;.中山大学药学院,广州 510006;.佛山市顺德区北滘医院,广东佛山 5811)
僵蚕来源于蚕蛾科昆虫家蚕Bombyx moriLinnaeus 4~5龄的幼虫感染(或人工接种)白僵菌Beauveria bassiana(Bals.)Vuillant而致死的干燥体,具有催眠、抗惊厥、息风止痉、清热解毒、祛风化痰及散结等功效[1]。而相关研究表明,僵蚕抗惊厥作用的主要有效成分是其体表白色粉末中含有的草酸铵[2]。不同的产地、不同的炮制方法以及白僵菌的繁殖条件都会影响僵蚕中草酸铵的含量[3]。目前,有关文献报道的关于僵蚕中草酸铵含量测定的方法主要有高效液相色谱法[4-5]、高效毛细管电泳法[6]、示波电位滴定法[7]、高锰酸钾氧化还原滴定法[8]等。高效液相色谱法的应用范围广、分离效率高、定量效果好,为目前最为常用的分析方法,但其流动相消耗大,且多为有毒的有机溶剂,对操作者和环境有一定的危害,另分析成本相对较高。而高效毛细管电泳法虽然需样量少、灵敏度高,但是毛细管容易损坏。示波电位滴定法是将高锰酸钾法与示波器结合起来指示终点的方法,该方法比高锰酸钾氧化还原滴定法更为直观灵敏、简便快速,但是滴定结果专属性差,准确性易受温度、酸度、滴定速度的影响。使用微流控芯片非接触电导检测的方法目前尚未见报道。
笔者建立了采用微流控芯片非接触电导法测定僵蚕中草酸铵的含量。该方法仪器集成程度高、操作简单,使用具有强分离效能的微流控芯片进行分离,而所用的非接触式电导检测避免了电极与溶液接触,寿命长,成本低,灵敏度较高,为僵蚕的质量控制提供一种高效、准确的分析方法。
1 材料
PMMA(Polymethyl methacrylate,聚甲基丙烯酸甲酯)十字通道芯片(微通道上宽30 μm,下宽100 μm,深30 μm,进样通道为十字结构,分离通道长44 mm,有效分离长度43 mm。大连理工大学微系统研究中心提供);非接触电导检测器(两电极间的距离0.6 mm。中山大学药学院提供)[9];微型压电陶瓷高压电源(中山大学药学院提供)[10];SHZ-D(Ⅰ)型循环水式真空泵(巩仪市英峪予华仪器厂);色谱工作站(中山大学医药仪器与应用研究所),数据记录与处理在普通P4微机上完成。
僵蚕(购自北京同仁堂广州药业连锁有限公司,产地:四川,批号:20140702、20140703、20140704),经中山大学姚美村副教授鉴定为蚕蛾科昆虫家蚕Bombyx moriLinnaeus 4~5龄的幼虫感染白僵菌Beauveria bassiana(Bals.)Vuillant而致死的干燥体;草酸铵对照品(广州化学试剂厂,分析纯,纯度≥99.5%);2-(N-吗啉代)乙磺酸(MES,上海晶纯试剂有限公司,超级纯);三羟甲基氨基甲烷(Tris,Farca Chemical Supplies,优级纯);其他试剂均为国产分析纯,水为二次蒸馏水。所有试剂使用前均经0.22 μm的微孔滤膜过滤。
2 方法与结果
2.1 试验条件
缓冲体系:3 mmol/L Tris+3 mmol/L柠檬酸(pH=3.0);分离电压:2.0 kV;进样时间:10.0 s;不加添加剂;非接触电导检测器激发电压:60 V(Vp-p);频率:60 kHz。
2.2 溶液的制备
2.2.1 对照品溶液 准确称取草酸铵对照品0.010 0 g,加水溶解并定容于10 ml量瓶中,得质量浓度为1.00 mg/ml的对照品贮备液,于4 Ⅰ下保存。临用前用缓冲溶液稀释成各浓度的对照品溶液。由于对照品溶液中所含的缓冲溶液配比与电泳介质的配比相同,因此可以消除背景干扰。进样前经0.22 μm的微孔滤膜过滤。
2.2.2 供试品溶液 取僵蚕样品适量,研碎,取约1.0 g,精密称定,将其转移至50 ml锥形瓶中,加入30 ml二次蒸馏水,后将锥形瓶放入50 Ⅰ恒温水浴锅中加热2 h,并不时用玻璃棒进行搅拌。然后过滤,滤渣加入少量二次蒸馏水继续加热提取,充分提取僵蚕中的草酸铵,得到的待测样品溶液再用滤纸过滤至100 ml量瓶中,定容,于4 Ⅰ下保存备用,作为供试品贮备液。临用时用缓冲溶液稀释10倍,作为供试品待测液,使得待测液中的缓冲溶液配比与电泳介质中的相同,消除背景干扰。进样前经0.22 μm的微孔滤膜过滤。
2.3 缓冲溶液种类
考察不同种类缓冲溶液对草酸铵分离检测的影响。在NaH2PO4-Na2HPO4、NaH2PO4-硼砂、柠檬酸-柠檬酸钠缓冲溶液中,草酸铵不出峰;在Tris-硼酸缓冲溶液中,虽然基线较为稳定,草酸铵出峰明显,但峰形较差,且为3个峰(如图1中A图所示);在醋酸-醋酸钠缓冲溶液中,草酸铵出峰明显、峰形较好,但基线不稳定(如图1中B图所示);在Tris-柠檬酸缓冲溶液中,基线稳定,基线噪音较低,草酸铵峰响应高、峰形较好、出峰时间短,是较理想的缓冲溶液(如图1中C图所示)。因此,优化选择Tris-柠檬酸作为缓冲溶液。
2.4 线性关系考察和检测限
按“2.2.1”项下方法配制质量浓度为1.0~200 μg/ml的系列对照品溶液,按“2.1”项下试验条件进行测定,记录峰面积。以质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得草酸铵的回归方程为y=0.755 4x+2.037(r=0.999 8)。结果表明,草酸铵质量浓度在10~150 μg/ml范围内与峰面积呈良好的线性关系。检测限为3.0 μg/ml(以信噪比为3计算)。
2.5 精密度试验
准确吸取1.00 mg/L的草酸铵对照品溶液5.00 ml,加入缓冲溶液稀释100倍,得50 μg/ml的草酸铵对照品溶液,按“2.1”项下试验条件重复进样6次,记录峰面积。结果,草酸铵峰面积的RSD为1.6%,表明仪器精密度良好。
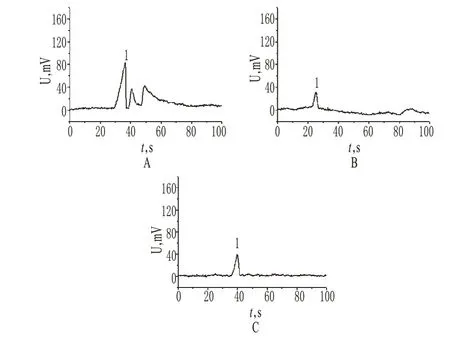
图1 草酸铵在不同缓冲溶液中的芯片毛细管电泳色谱图A.Tris-硼酸;B.醋酸-醋酸钠;C.Tris-柠檬酸;1.草酸铵Fig 1 Microchip capillary electropherograms of ammonium oxalate in different buffer solutionsA.Tris-H3BO3;B.HAc-NaAc;C.Tris-citric acid;1.ammonium oxalate
2.6 稳定性试验
取样品(批号:20140702)适量,按“2.2.2”项下方法制备供试品待测液,4 Ⅰ下保存,分别于放置0、2、4、6、8 h时按“2.1”项下试验条件进样测定,记录峰面积。结果,草酸铵峰面积的RSD为1.8%,表明供试品待测液在8 h内稳定性良好。
2.7 重复性试验
精密称取同一批次的样品(批号:20140702)适量,共6份,精密称定,按“2.2.2”项下方法制备供试品待测液,再按“2.1”项下试验条件进样测定,记录峰面积。结果,草酸铵峰面积的RSD为1.9%,表明该方法重复性良好。
2.8 加样回收率试验
精密量取“2.2.2”项下供试品贮备液1 ml(批号:20140702),共9份,分别置于10 ml量瓶中,加入草酸铵对照品(1.00 mg/ml),然后加入适量的缓冲溶液,定容至10.0 ml。按“2.1”项下试验条件进行测定,计算加样回收率,结果见表1。
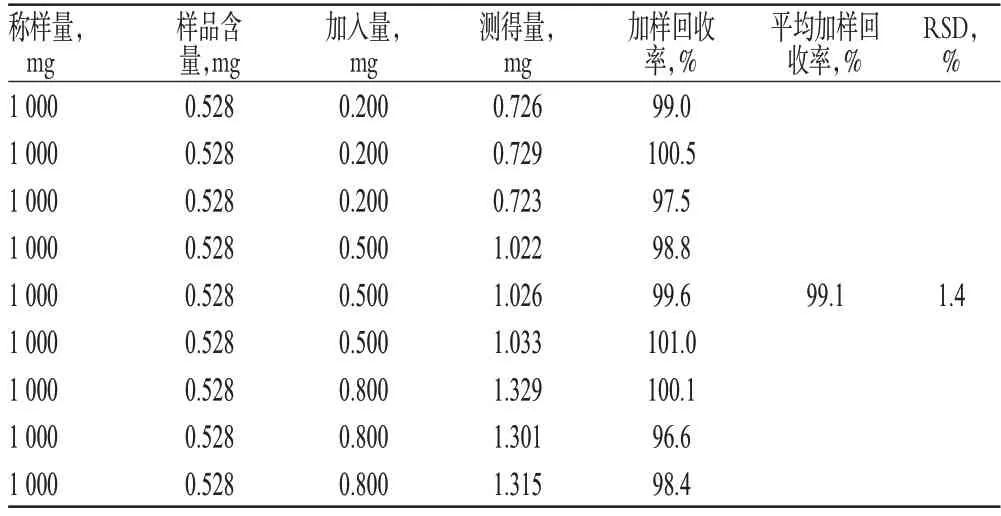
表1 加样回收率试验结果(n=9)Tab 1 Results of recovery tests(n=9)
2.9 样品含量测定
取“2.2”项下供试品待测液和对照品溶液(经缓冲溶液适当稀释)适量,按“2.1”项下试验条件进样测定,记录电泳图,见图2。
取3批样品(批号:20140702、20140703、20140704),按“2.2.2”项下方法制备供试品待测液,按“2.1”项下试验条件进样测定,记录峰面积,代入回归方程计算出草酸铵的质量浓度,从而得出其质量分数,结果见表2。
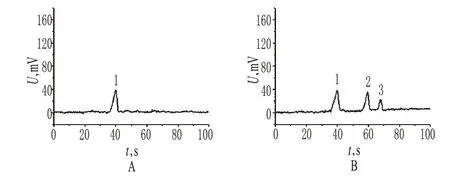
图2 芯片毛细管电泳谱图A.对照品;B.供试品;1.草酸铵;2、3.未知物Fig 2 Microchip capillary electropherogramsA.reference;B.test samples;1.ammonium oxalate;2-3.unknown impurities
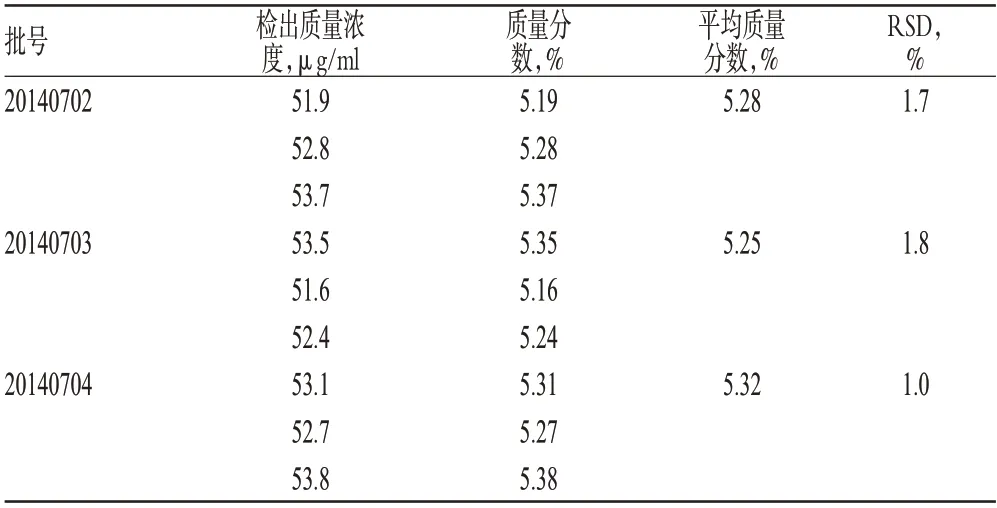
表2 样品含量测定结果(n=3)Tab 2 Results of content determination of samples(n=3)
3 讨论
3.1 缓冲溶液
草酸铵在各种不同缓冲溶液的出峰情况结果见“2.3”项。草酸铵在NaH2PO4-Na2HPO4等缓冲溶液中不出峰,可能与缓冲溶液的电导与样品的电导相差不大有关;而在Tris-硼酸缓冲溶液中分为3个峰,可能是由于草酸铵与缓冲溶液中的硼酸生成了配合物;在醋酸-醋酸钠缓冲溶液中,基线不稳定,可能是由于缓冲溶液离子强度过大。因此,最后优化选择Tris-柠檬酸作为缓冲体系。
除了种类,缓冲溶液中两组分比例和浓度对分离检测也有较大的影响。笔者分别考察了Tris(2~5 mmol/L)和柠檬酸(2~5 mmol/L)不同浓度配比的缓冲体系对分离检测的影响。结果发现,当两组分中Tris配比低时,峰形出现拖尾,可能是因为低离子强度的缓冲液造成了样品的吸附;当Tris配比高时,峰形得到改善,但灵敏度下降;当柠檬酸配比高时,基线噪音增加。结果显示,两者比例为1∶1时,峰形与基线均较好。因此,优化Tris和柠檬酸比例为1∶1。试验发现,随着Tris和柠檬酸总体浓度的增大,电流也随着增大,焦耳热效应使基线噪声也随之增加,影响分离检测效果。因此,综合考虑基线噪音及稳定性、出峰情况等分离检测效果,优化选择3 mmol/L Tris+3 mmol/L柠檬酸(pH=3.0)作为缓冲体系。
3.2 添加剂
一般情况下,向背景电解质中加入添加剂可影响电渗流、迁移率和控制焦耳热等,从而改善电泳分离分析效果。试验中考察了乙醇、甲醇、丙酮、丙二醇、β-环糊精(β-CD)、十二烷基磺酸钠(SDS)等几种添加剂不同添加量对分离检测效果的影响。结果显示,加入乙醇(1%~5%,体积分数,下同)、甲醇(1%~5%)以及丙酮(1%~5%)后,虽然草酸铵峰形变好,但基线漂移,这可能是由于醇类和酮类物质具有挥发性,从而导致基线上飘;加入丙二醇(1%~5%)后,峰形变差,重复性不好,而且基线不稳定,这可能是丙二醇的黏度大及其吸湿性造成;加入β-CD(5%~10%)和SDS(5%~10%),对分离检测无改善,且基线不稳定,噪音增大。综合上述检测效果,优化选择缓冲溶液中不加添加剂。
3.3 进样时间
笔者又考察了进样时间为5.0~20.0 s时对草酸铵分离和检测的影响。随着进样时间的延长,峰高与峰面积呈增大趋势;但当进样时间超过10.0 s后,峰高增加的幅度减少,这可能是由于进样时间过长,进样量超出了芯片检测的载样量;而且随着进样时间的增加,峰出现展宽和拖尾。综合考虑,优化选择进样时间为10.0 s。
3.4 分离电压
在微流控芯片系统中,分离电压的大小可以直接影响电场强度的大小,从而影响物质的迁移速度。而增加组分的迁移速度是减少谱带展宽、提高分离效率的重要途径。因此,本研究考察了分离电压1.0~2.5 kV对样品检测效果的影响。结果发现,随着分离电压的增加,峰高逐渐增加,峰形尖锐,出峰时间缩短,信噪比增加。但是,过高的分离电压可产生较大的电流,使电泳体系产生大量的焦耳热,基线噪音随之增大、信噪比下降,从而影响检测效果。综合考虑峰形、灵敏度、噪音以及出峰时间等因素,优化选择分离电压为2.0 kV。
综上所述,该方法简便、快速、重复性好,为僵蚕的质量控制提供了一种新方法。
[1]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:352.
[2]徐冲,商思阳,刘梅,等.僵蚕化学成分和药理活性的研究进展[J].中国药房,2014,25(39):3 732.
[3]赵清,徐月清,冯天铸,等.不同炮制方法对僵蚕指标性成分的含量影响研究[J].时珍国医国药,2011,22(3):657.
[4]张琦,严铸云,宋杰,等.HPLC法测定不同产地僵蚕中草酸铵含量[J].现代生物医学进展,2008,8(5):915.
[5]彭新君,许光明,李明娟,等.高效液相色谱法测定僵蚕中草酸铵的含量[J].中南药学,2006,4(4):255.
[6]黄盖群,刘刚,江生,等.高效毛细管电泳法测定中药材白僵蚕抗惊活性成分方法的探讨[J].西南农业学报,2008,21(3):849.
[7]丁亚平,吴庆生.示波电位滴定法测定僵蚕中草酸铵[J].分析试验室,1997,16(5):14.
[8]彭新君,彭延古,曾序求,等.僵蚕提取液中蛋白质和草酸铵等成分的定量分析[J].中国中医药信息杂志,2005,12(9):39.
[9]Chen Z,Li Q,Li O,et al.A thin cover glass chip for contactless conductivity detection in microchip capillary electrophoresis[J].Talanta,2007,71(5):1 944.
[10]陈缵光,王立世,莫金垣.微全分析系统专用微型电源的研制[J].高等学校化学学报,2004,25(增刊):26.
猜你喜欢
四川蚕业(2022年1期)2022-06-06
四川蚕业(2021年4期)2021-03-08
肾脏病与透析肾移植杂志(2021年1期)2021-01-13
化学与粘合(2020年6期)2020-03-08
四川蚕业(2020年4期)2020-02-10
科技视界(2018年30期)2018-01-31
科技视界(2017年25期)2017-12-11
中成药(2017年8期)2017-11-22
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
