
基于单克隆抗体的肿瘤免疫疗法研究进展
2015-03-08 06:28郭亚军
生物工程学报 2015年6期
郭亚军
“免疫逃逸”是肿瘤发生过程中的重要标志和主要机制。肿瘤的免疫疗法 (Immunotherapy)是通过增强机体对癌细胞的识别能力,利用机体自身的免疫能力对肿瘤进行清除[1]。与手术、化疗、放疗等传统疗法相比,肿瘤免疫疗法具有肿瘤靶向性好、疗效持久,临床副作用小等优点。目前,应用于临床的肿瘤免疫疗法主要有:细胞疗法 (ProvengeTM)、细胞因子 (IL-2,IFN-α等) 和单克隆抗体。其中单克隆抗体的应用最为广泛。自 1997年首个抗肿瘤抗体药物Rituximab上市以来,目前已有 20余种单抗药物用于多种血液肿瘤、实体瘤的治疗。
近两年,又不断有新型结构类别的抗体药物进入临床。如:靶向免疫检查点的拮抗型抗体,能够招募 T细胞的双特异性抗体 (Bi-specific T-cell engaging antibodies, BiTE),以及负载有小分子化药的抗体药物偶联物 (Antibody drug conjugates, ADCs) 等。上述新一代抗体药物在多种恶性肿瘤治疗上均取得了突破性进展,因此,2013年Science评选的“年度十大科学突破”中,以抗CTLA-4抗体Ipilimumab为代表的“肿瘤免疫疗法”位列榜首[2]。
1 抗肿瘤抗体药物的发展现状
1.1 抗体疗法在肿瘤治疗上的应用
1897年,Paul Ehrlich在著名的“魔术子弹”学说,首次指出靶向病原相关抗原的抗体可以用来治疗疾病[3]。20世纪60年代,基于血清学研究众多肿瘤组织特异性抗原被发现,并成为了日后抗肿瘤抗体药物开发的重要靶点。1975年“杂交瘤”技术的出现[4],使得单克隆抗体的大规模制备成为可能。1980年,靶向淋巴细胞抗原的单抗 AB89静脉注射到非霍金奇淋巴瘤患者体内后,肿瘤细胞得到暂时的抑制[5],这是临床上最早关于单抗治疗肿瘤的记载。
1997年靶向B细胞抗原CD20的单抗药物Rituximab进入临床,并在治疗非霍金奇淋巴瘤上获得成功[6]。此后,Cetuximab、Trastuzumab、Bavacizumab等治疗性抗体陆续进入临床,截止2014年12月用于癌症治疗的抗体药物总计25种 (表1)。其中,据FiercePharma统计2013年销售额排名前三的抗肿瘤抗体药物为:Rituximab (75亿美元),Bevacizumab (67亿美金),Trutuzumab (65 亿美金)。
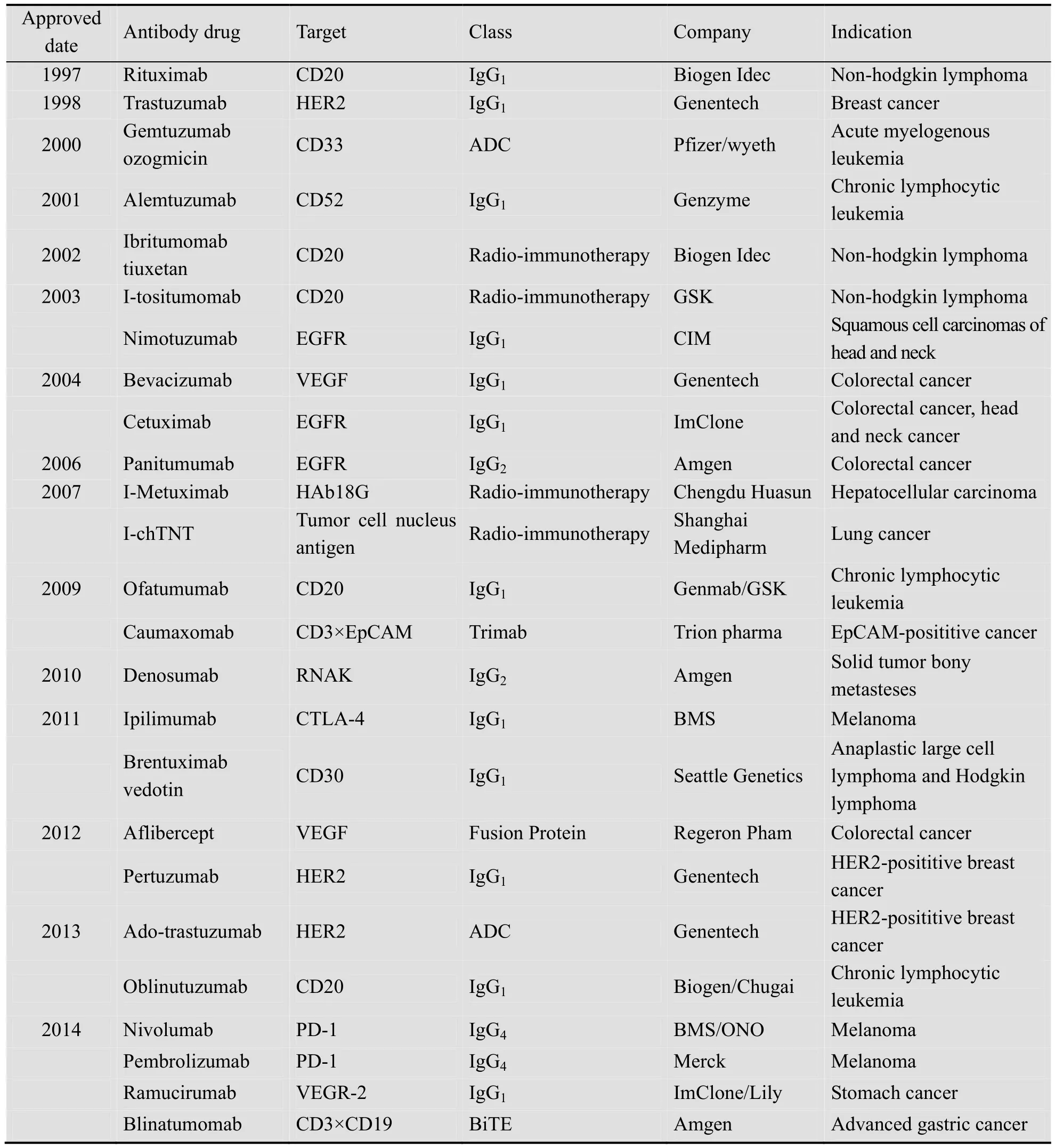
表1 用于治疗肿瘤的单克隆抗体药物 (数据截止到2014年12月)Table 1 The therapeutic antibody for cancer treatment (date: December 2014)
除此之外,尚有 160多种抗肿瘤抗体药物处于临床研究阶段,药物靶点涵盖血液分化抗原 (CD20、CD30、CD33和CD52等),细胞生长因子 (CEA、EGFR、HER2、HER3、MET和IGFR1等),肿瘤坏死因子配基 (TRAIL-R1、TRAIL-R2和 RANKL等),血管内皮生长因子(VEGF) 等[7]。
1.2 重组抗体药物的发展趋势
1983年最早上市的治疗性抗体OKT-3为鼠源抗体,由于鼠源序列在人体内引起“人抗鼠抗体”,此后为减少鼠源抗体的免疫原性,出现了“嵌合抗体”、“人源化抗体”等技术。近年来,可利用体外展示技术 (噬菌体、核糖体、酵母菌等) 或直接免疫商品化转基因小鼠(XenoMouseTM、HuMabTM、VelociMouseTM等)获得全人源抗体。
此外,也可通过免疫其他物种获得“成药性”更好的单抗。如:羊驼抗体结构天然缺失轻链分子量小被称为“纳米抗体”[8];利用兔杂交瘤技术获得的兔单抗,识别抗原表位多,易于人源化等[9];免疫鲨鱼获得的单抗具有热稳定性好、耐酸性强等优点[10]。
1.3 抗体药物治疗肿瘤的作用机制
临床上应用广泛的抗肿瘤抗体药物,其作用机制为通过靶向肿瘤细胞表面抗原,阻断肿瘤生长因子信号通路,或遏制肿瘤微环境中新生血管的形成。同时,抗体恒定区介导的ADCC(Antibody-dependent cellular cytotoxiciy)、CDC(Complement-dependent cytotoxicity)、ADPC(Antibody dependent cellular phagocytosis) 等效应功能可进一步杀伤肿瘤细胞[11-12]。近年的研究发现,对于Trastuzumab[13]与Cetuximab[14]而言,激活体内效应 T细胞也是抗体药物杀伤肿瘤的重要机制。
值得注意的是,针对同一靶点的不同单抗药物,其作用机制并不一定完全相同,临床疗效与不良反应也有所差异[15]。如:Trastuzumab和Pertuzumab均靶向HER2,但是两者的抗原识别表位不同,前者可抑制HER2受体的同源二聚化和异源二聚化,而后者只抑制HER2与EGFR或HER3的异源二聚化[16],因此两者在临床上联合用药具有“协同效应”;靶向EGFR的Cetuximab、Panitumumab和Nimotuzumab虽然抗原结合表位相同,但是由于亲和力和IgG亚型的差异,临床上引起的皮肤毒性也有所差异[17];Ofatumumab和Rituximab均靶向B细胞抗原CD20,但是由于 Ofatumumab的结合表位不同和解离速度较慢,其在体外介导的效应功能更强[18]。
2 新型抗肿瘤抗体的药物靶点与作用机制
近年来,随着“抗体工程”技术的发展以及免疫检查点药物靶点的发现,临床上已经出现新一代抗肿瘤抗体,其作用机制和临床疗效显著区别传统抗体药物。这其中最具代表性的当属靶向免疫检查点蛋白的共抑制因子拮抗剂、可同时靶向不同抗原的多特异性抗体以及负载有高杀伤力化学药物的ADCs药物。
2.1 靶向免疫检查点的抗体药物
T细胞表面的共刺激因子通过与相应配基结合提供激活细胞免疫的“第二信号”,免疫检查点 (Immune checkpoint) 蛋白正是体内效应细胞激活所必需的共刺激因子。作为药物靶点的免疫检查点蛋白,主要属于免疫球蛋白超家族 (如:CTLA-4、PD-1等) 和肿瘤坏死因子超家族 (如:4-1BB、OX40、GITR等)。通过设计“共抑制因子”的拮抗剂或“共刺激因子”的激动剂,可以启动机体的细胞免疫治疗恶性肿瘤[19]。
2012年靶向 CTLA-4的单抗药物Ipilimumab[20]在美国上市,用于治疗晚期黑色素瘤。2014年靶向 PD-1的 Nivolumab[21]、Pembrolizumab[22]已经先后在日本、美国上市;靶向PD-L1抗体MPDL3820A成为30年来对转移性膀胱癌唯一有效的抗体药物[23]。此外,靶向共刺激因子4-1BB的激动剂型抗体Urelumab和PF05082566,以及靶向CD40激动剂型抗体的Lucatumumab和Dacetuzumab也已进入临床研究阶段[24]。
2.2 多特异性抗体
传统抗体药物通过封闭单一信号通路抑制肿瘤生长,临床上易出现单抗药物的耐药性。而多特异性抗体通过靶向不同抗原同时阻断多个信号通路,在临床应用上更具优势。如:MM-111通过同时靶向HER2/HER3,有效抑制调蛋白诱导的 HER3信号通路激活[25-26];同时靶向VEGF-A和Ang-2的CrossMab双功能抗体(Ang-2-VEGF-A) 有效阻断肿瘤的血液传播[27];封闭EGFR/HER2/HER3/VEGF的四特异性抗体在体内对于HER2耐药株有很好的抑瘤效果[28]。
另外,多特异性抗体还可通过靶向效应细胞 (T细胞、NK细胞等) 抗原,利用效应细胞对肿瘤进行杀伤。2009年三特异性抗体Caumaxomab在欧盟上市,该抗体同时靶向肿瘤抗原 (EpCAM) 和 T细胞抗原 (CD3),依靠招募 T细胞和恒定区介导的效应功能等机制杀伤肿瘤[29]。今年在美国上市的 BiTE 药物Blinatumomab,由靶向效应T细胞 (CD3) 和肿瘤细胞抗原 (CD19) 的单链抗体串联而成,用于治疗急性淋巴性白血病。由于 BiTE药物分子量小(55 kDa),能够充分激活T细胞,剂量可减少到普通抗体药物的1/1 000 (0.005 mg/m2)[30]。但是,由于BiTE抗体缺乏恒定区,存在体内半衰期较短的缺点,临床给药需借助体内微型泵。此外,还有靶向 CD16A和 CD30的双特异性抗体(TandAb) 通过招募NK细胞对肿瘤细胞进行杀伤[31]。
2.3 抗体化学药物偶联物
ADC药物由靶向肿瘤的抗体药物与高杀伤力小分子化药偶联而成,借助抗体实现化药对肿瘤组织的靶向递送。这种设计策略既可提高抗体药物的杀伤能力,又可提高小分子化药的治疗窗。目前,已经有 3种抗体药物偶联物获批上市,尚有超过30种ADC药物处于临床研究阶段[32]。
惠氏公司开发的 Gemtuzumab ozogamicine(MylotargTM) 是抗CD33单抗联有卡其霉素,该药2001年获FDA批准用于治疗复发性急性髓系淋巴瘤[33],2010年由于其有效性与安全性受到质疑自动退市[34]。西雅图基因公司开发的Brentuximab Vedotin (SGN-35),是利用靶向CD30的嵌合抗体偶联毒素MMAE[35],2011年获 FDA批准用于在治疗复发性霍奇金氏淋巴瘤、大细胞淋巴瘤等[36];基因泰克公司开发的Dotrastuzumab Emtansine (KadcyclaTM) 则是Trastuzumab联有化药DM1,用于治疗HER2阳性乳腺癌。
3 新型抗肿瘤抗体的设计策略
上述新型抗体药物的出现,很大程度得益于近年来“抗体工程”技术的发展[37]。在此基础上,一方面不断有针对原有靶点的第二代抗体药物出现,如:靶向 CD20的GA101经过糖基化改造,其效应功能较原型抗体Rituximab提高近百倍。另一方面,抗体工程技术直接促成了新型结构抗体药物进入临床,如:利用“四杂交瘤”技术表达双特异性抗体;利用巯基偶联、氨基偶联技术合成ADC药物;利用肿瘤部位特异酶解肽构建的抗体“前药”(Probody) 等[38]。
3.1 双特异性抗体的构建方法
通过对抗体结构的改造或可变区的突变,可以构建靶向两 (多) 个抗原的双 (多) 特异性抗体。由于双特异性抗体构建方式灵活,目前已有报道的多特异性抗体形式至少45种[39]。按照其结构特征可分为:IgG对称型、IgG非对称型、IgG/Fc融合蛋白和单链抗体等类别,常见的双特异性抗体形式 (图1)[40]。
对称型IgG多功能抗体由于两条重链、轻链各自相同,表达、纯化工艺相对简单最具应用前景。如:DVD-IgG是在IgG抗体可变区的轻、重链上,分别串联上靶向另一抗原的轻、重链可变区[41];“Two in One”型抗体是利用体外展示技术直接筛选到可同时靶向不同抗原的抗体,利用该技术获得了可同时结合VEGF和HER2的高亲和力抗体[42];利用酵母菌展示文库,对IgG的恒定区进行突变,在不改变恒定区效应能力、半衰期的前提下,获得能够与抗原高亲和力结合的恒定区,此类抗体的可变区与恒定区均可结合不同抗原,称之为“mabs2”抗体[43]。
非对称型 IgG抗体通常的两条重链来自靶向不同抗原的抗体,需要进行重新配对组装。这种组装一般是基于在重链CH3区的互补氨基酸序列,如:可在重链CH3区构建“纽扣结构”(Knobs-into-holes)[44]、“ 静 电 吸 引 ”(Electrostatically-matched)[45]、“链交换结构域”(Strand exchange engineered domain,SEED)[46]等互补结构域;或CH3区添加可去除的“亮氨酸拉链”(LUZ-Y)[47],来提高不同重链间的配对效率。非对称型 IgG抗体的轻链一般使用“通用轻链”或利用“cross over”[48]进行构建。或通过在抗体骨架上化学连接不同的抗原识别肽,构建“CovX-Bodies”形式的双功能抗体,利用该技术构建的同时靶向VEGF和Ang2的双功能抗体已经进入临床研究阶段[49]。
“dock and lock”技术利用蛋白激酶A的调节亚基和A激酶锚定蛋白的锚定区域的特异性结合能力,设计二聚体对接结构域连接两个不同靶向的可变区[50],也可以直接串联靶向不同抗原的可变区构成“Diabody”抗体[39],或通过二硫键连接进一步稳定结构构成“DART”(Dual affinity retargeting technology) 抗体[51]。此类抗体由于缺乏恒定区,会导致半衰期短、无Fc效应功能等,但是由于其分子量小更具肿瘤穿透力。
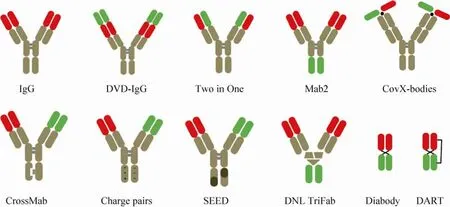
图1 常见的双特异性抗体结构[40]Fig.1 Structure of bi-specific antibody[40].
3.2 改变恒定区的效应功能和半衰期
不同的抗体亚型其效应功能有所不同,一般来说,抗体亚型的效应功能遵循着下述规律:IgG3>IgG1>>IgG2≈IgG4,此外,由于 IgG3存在半衰期较短,IgG4易在体内形成半抗体,两者成药性较差[52]。所以早期的靶向肿瘤的抗体多选择IgG1型,而靶向免疫细胞的抗体药物多选择 IgG4突变型 (S228P) 或无效应功能的 IgG1突变体 (N297A)。
抗体恒定区的糖基化程度对其效应功能影响显著。降低岩藻糖修饰程度,可提高Rituximab的 ADCC效应近百倍[53];为进一步提高 IgG1的CDC功能,可利用IgG3的恒定区构建IgG1/3嵌合型抗体[54]。此外,通过对恒定区进行定点突变 (G236A),通过提高与巨噬细胞 FcγIIa受体的亲和力,提高抗体介导“调理效应”[55]。针对IgG3半衰期较短的问题,可对恒定区进行定点突变提高与 FcRn的亲和力,延长 IgG3的半衰期[56-57]。
对于不需要效应功能的中和抗体,通常选择效应功能较弱的突变型IgG4(S228P),该突变体可有效避免“半抗体”的形成[58];或者构建没有效应功能的IgG2/4嵌合型抗体[59]。
3.3 化学药物与抗体的偶联技术
在 ADCs药物的设计上,药物靶点一般选择肿瘤表面特异性表达,且可介导内吞作用的抗原。偶联的小分子化药对肿瘤细胞应具有较高的杀伤能力,如:常用的微管抑制剂MMAE和DM1其IC50低于nmol/L级。化药与抗体通过连接子 (Linker) 连接,连接子在胞内的溶酶体内经酸性环境、还原环境或经蛋白酶切后水解进而释放药物。传统的ADC连接方式,采用氨基偶联或巯基偶联,由于抗体药物平均含有40个赖氨酸残基、8个半胱氨酸,这种非特异性偶联方式下 ADC的“抗体药物比率”(Antibody drug ratio,ADR) 为 0−8。由于 ADR异质性会造成ADC药物在临床上药代动力学和药效学上的差异。因此,开发位点特异性连接的ADCs是此技术领域的发展趋势[60]。
通过在可变区插入高活性巯基(PHESELECTOR) 在不影响可变区结合活性的前提下,可实现ADC药物的位点特异性偶联。基于此技术的“THIOMAB”抗体偶联物,分子位点特异性连接2个小分子化药,体内的“治疗窗口”较非特异性连接ADC药物大为提高[61-62];此外,也可通过在抗体序列中插入高活性的非天然氨基酸 (如:硒代半胱氨酸[63]、乙酰半胱氨酸[64]),或借助糖基转移酶[65]和转谷氨酰胺酶[66]来实现化药与抗体分子的位点特异性偶联。
4 抗体肿瘤免疫疗法引起的药物不良反应
抗体药物由于临床剂量大 (5−10 mg/kg),给药方式多通过静脉滴注,因此临床上最为常见的药物不良反应是,因药物免疫原性引起的“急性输液反应”。此外,由于药物靶点在正常组织上的非特异性表达,抗体药物还会在临床上出现“On-target”毒性。如:靶向 EGFR的Panitumumab和Cetuximab易引起皮肤毒性;靶向 HER2的 Herceptin易引起心脏毒性;靶向VEGF抗体Bevacizumab会增加血栓栓塞风险;靶向CD52的单抗的Alemtuzumab可引起血小板减少;靶向CD20的Rituximab会增加病毒感染风险等[67]。
4.1 “细胞因子风暴”
靶向T细胞共刺激因子 (CD3和CD28等)受体的激动型抗体,由于可以充分激活 T细胞释放炎症因子,在临床上也会引起严重的“细胞因子风暴”。如:在 CD28激动剂 TGN1412在临床Ⅰ期试验中,6名健康志愿者按0.1 mg/kg静脉给药后,均发生了严重的系统性炎症反应[68]。但是,当 TGN1412的给药剂量降至5 μg/kg以下后,未出现明显的细胞因子释放症状,目前该药重新进入临床研究[69]。
另外,双特异性抗体Blinatumomab由于靶向T细胞抗原CD3,激活T细胞释放细胞因子后会进一步激活巨噬细胞,导致患者会出现“巨噬细胞综合症”。一般临床上通过使用抗炎症药物Tocilizumab缓解不良反应[70]。
4.2 免疫相关不良反应
免疫检查点蛋白通过与其配基结合抑制 T细胞活化,维持着机体正常组织的免疫耐受。靶向免疫检查点蛋白的抗体药物在激活免疫细胞的同时,也会导致患者出现免疫相关不良反应 (Immune-related adverse events),而且不良反应的严重程度往往和治疗效果正相关。对此临床上一般使用抗炎症激素类药物进行治疗。如:Ipilimumab在给药过程中常出现轻度到中度不良反应,也有部分患者出现持续腹泻、致命的表皮坏死松解症等[71-72]。抗PD-1单抗由于其作用机理在于激活“耗竭”效应细胞,临床副作用相对较小,主要表现为疲劳、皮疹和肠炎等,极少数患者出现肺炎症状,而且给药终止或使用抗炎激素后不良反应即可消失[21]。
4.3 ADCs药物的不良反应
虽然,ADCs药物借助单抗实现小分子化药的靶向递送,其药物“治疗窗口”已经较传统化药有了大幅提高。但是,由于小分子脱落或药物靶点的非组织特异性表达,在临床上仍存在着一定的毒副作用。如:2004年上市的ADCs药物 Gemtuzumab ozogamicin被认为可能引起致命性的“肝静脉阻塞病”(Hepatic venoocclusive disease)[73],经风险收益评估后该药于2010年撤市。另外,最新研究表明Brentuximab vedotin在临床上会引起严重的胰腺炎[74]。
5 国内抗体药物研究进展
近年来,我国学者在抗体药物的基础研究及产业化进程中均取得了长足的进展。如:在抗原-抗体晶体结构解析方面,中国科学院丁建平课题组成功解析出抗IL-2单抗Dalizumab[75]、抗 CD20单抗 Rituximab[76]、抗 LFA-1单抗Efalizumab[77]等多个抗体药物的晶体结构;清华大学饶子和院士课题组在解析出 Infliximab[78]晶体结构的基础上,对靶向TNFα的Infliximab和 Adalimumab的分子作用机制进行了深入探讨[79]。基于上述抗体结构的基础研究,我国学者尝试利用计算机辅助设计技术,通过定点突变进一步提高抗体与靶标的亲和力。如:第二军医大学李博华等对 Rituximab进行三点突变(H57DE/H102YK/L93NR) 后,克服了Ⅰ型抗CD20抗体不能诱导细胞凋亡的缺点[80]。军事医学科学院沈倍奋院士课题组利用计算机辅助设计的抗 TNF-a拮抗剂 ATD5,能够有效地抑制TNF-a与其受体TNR-1/2的结合[81]。
在新型抗体设计方面,军事医学科学院郭宁教授课题组成功构建了抗HER2与CD16的双功能抗体[82]。第二军医大学李博华等利用Trastuzmab和Pertuzumab可变区所构建的双功能抗体,能够有效抑制 ErbB2的“异二聚体”化,该抗体对于 Trastuzumab耐药株治疗有效[83];寇庚等构建靶向VEGF和骨桥蛋白的双功能蛋白[84],张大鹏等构建可同时结合VEGF-A和VEGF-C的融合蛋白[85],均能有效抑制肿瘤血管新生,在体内显示出较好抑瘤效果。Li等构建的新型四价抗CD20抗体具有CDC、ADCC及诱导细胞凋亡等多种作用机制[86]。此外,在抗体药物偶联物研究方面,浙江大学陈枢青课题组在ADC药物的基础上提出“XDC”概念,其设计的 TRAIL-MMAE偶联药物进入肿瘤细胞后,经组织蛋白酶降解后定点释放毒素MMAE[87]。
在抗体药物产业化方面,目前我国生产企业已经有多个治疗性抗体药物上市,其中包括靶向 TNF-a融合蛋白的“益赛普”、“强克”等[88],靶向EGFR的尼妥珠单抗[17]等。2014年我国自主研发的新型VEGF受体融合蛋白“康柏西普”(Conbercept),可同时结合 VEGF-A、VEGF-B和PIGF等多个配基[89],目前已经获批上市用于治疗湿性老年黄斑变性。
6 展望
2014年,抗PD-1抗体在临床上取得的重大突破,Nivolumab和Pembrolizumab先后在日本和美国上市,同时抗PD-L1抗体也成为近30年来唯一对晚期膀胱癌治疗有效的药物。但是临床上仍有70%的癌症患者对抗PD-1抗体治疗无效。进一步研究发现,肿瘤的免疫原性以及患者的临床免疫分型与治疗效果密切相关。肿瘤免疫原高、T淋巴细胞浸润型的患者往往对于抗PD-1抗体更为敏感。而非T细胞浸润型肿瘤患者使用PD-1抗体后往往治疗无效[90-91]。因此,靶向免疫检查点的抗体药物在临床使用中,通常会与放疗、化疗联用以提高肿瘤的免疫原性,或者与通过其他免疫疗法 (树突状细胞疫苗,细胞因子,细胞疗法等) 联用来充分激活机体的免疫功能。此外,作为以靶向性见长的抗体类药物,生物标记物 (Biomarker) 对于指导其临床用药尤为重要。如:Cetuximab在临床给药前需对病人KRAS基因进行筛分。而适用于抗PD-1抗体的临床标记物也正在研究中。因此,如何设计合理、有效的联合用药方案,以及寻找用于指导临床用药的生物标记物,将是未来基于抗体的肿瘤免疫疗法临床应用的发展方向。
[1] Hanahan D, Weinberg RA. Hallmarks of cancer:the next generation. Cell, 2011, 144(5): 646–674.
[2] Couzin-Frankel J. Cancer immunotherapy. Science,2013, 342(6165): 1432–1433.
[3] Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer, 2008, 8(6): 473–480.
[4] Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity.Nature, 1975, 256(5517): 495–497.
[5] Nadler LM, Stashenko P, Hardy R, et al.Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res, 1980,40(9): 3147–3154.
[6] Maloney GD, Grillo López AJ, White CA, et al.IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood, 1997,90(6): 2188–2195.
[7] Reichert JM, Dhimolea E. The future of antibodies as cancer drugs. Drug Discov Today, 2012,17(17/18): 954–963.
[8] Harmsen MM, de Haard HJ. Properties, production,and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol, 2007, 77(1):13–22.
[9] Feng LF, Wang X, Jin HC. Rabbit monoclonal antibody: potential application in cancer therapy.Am J Transl Res, 2011, 3(3): 269–274.
[10] Kovaleva M, Ferguson L, Steven J, et al. Shark variable new antigen receptor biologics-a novel technology platform for therapeutic drug development. Expert Opin Biol Ther, 2014, 14(10):1527–1539.
[11] Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer, 2012, 12(4): 278–287.
[12] Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol, 2010, 10(5):317–327.
[13] Park S, Jiang ZJ, Mortenson ED, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity.Cancer Cell, 2010, 18(2): 160–170.
[14] Yang XM, Zhang XM, Mortenson ED, et al.Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther,2013, 21(1): 91–100.
[15] Shim H. One target, different effects: a comparison of distinct therapeutic antibodies against the same targets. Exp Mol Med, 2011, 43(10): 539–549.
[16] Sakai K, Yokote H, Murakami-Murofushi K, et al.Pertuzumab, a novel HER dimerization inhibitor,inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci, 2007, 98(9): 1498–1503.
[17] Ramakrishnan MS, Eswaraiah A, Crombet T, et al.Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. mAbs,2009, 1(1): 41–48.
[18] Lin TS. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers Med,2010, 3: 51–59.
[19] Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer, 2012,12(4): 252–264.
[20] Hodi FS, O'Day SJ, McDermott DF, et al.Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med, 2010, 363(8):711–723.
[21] Topalian SL, Hodi FS, Brahmer JR, et al. Safety,activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med, 2012, 366(26):2443–2454.
[22] Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med, 2013, 369(2): 134–144.
[23] Powles T, Eder JP, Fine GD, et al. MPDL3280A(anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature, 2014,515(7528): 558–562.
[24] Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov, 2013, 12(2): 147–168.
[25] Robinson MK, Hodge KM, Horak E, et al.Targeting ErbB2 and ErbB3 with a bispecific single-chain Fv enhances targeting selectivity and induces a therapeutic effect in vitro. Br J Cancer,2008, 99(9): 1415–1425.
[26] McDonagh CF, Huhalov A, Harms BD, et al.Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3.Mol Cancer Ther, 2012, 11(3): 582–593.
[27] Kienast Y, Klein C, Scheuer W, et al.Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor,antiangiogenic, and antimetastatic efficacy. Clin Cancer Res, 2013, 19(24): 6730–6740.
[28] Hu S, Fu W, Xu W, et al. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3 and VEGF through disruption of HER/MET crosstalk. Cancer Res, 2015, 75(1):159-170.
[29] Seimetz D. Novel monoclonal antibodies for cancer treatment: the trifunctional antibody catumaxomab(removab). J Cancer, 2011, 2: 309–316.
[30] Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science, 2008,321(5891): 974–977.
[31] Reusch U, Burkhardt C, Fucek I, et al. A novel tetravalent bispecific TandAb (CD30/CD16A)efficiently recruits NK cells for the lysis of CD30+tumor cells. mAbs, 2014, 6(3): 728–739.
[32] Sassoon I, Blanc V. Antibody-drug conjugate(ADC) clinical pipeline: a review//Ducry L, Ed.Antibody-Drug Conjugates. Methods in Molecular Biology Volume 1045. Humana: Humana Press,2013: 1–27.
[33] Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res, 2001,7(6): 1490–1496.
[34] Castaigne S, Pautas C, Terré C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia(ALFA-0701): a randomised, open-label, phase 3 study. Lancet, 2012, 379(9825): 1508–1516.
[35] Younes A, Bartlett NL, Leonard JP, et al.Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med, 2010,363(19): 1812–1821.
[36] Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma:results of a phase II study. J Clin Oncol, 2012,30(18): 2190–2196.
[37] Beck A, Wurch T, Bailly C, et al. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol, 2010, 10(5):345–352.
[38] Desnoyers LR, Vasilijeva O, Richardson JH, et al.Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med, 2013, 5(207): 207ra144.
[39] Kontermann RE. Dual targeting strategies with bispecific antibodies. mAbs, 2012, 4(2): 182–197.
[40] Weidle UH, Kontermann RE, Brinkmann U.Tumor-antigen-binding bispecific antibodies for cancer treatment. Semin Oncol, 2014, 41(5):653–660.
[41] Wu CB, Ying H, Grinnell C, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol,2007, 25(11): 1290–1297.
[42] Bostrom J, Yu SF, Kan D, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science, 2009,323(5921): 1610–1614.
[43] Wozniak-Knopp G, Bartl S, Bauer A, et al.Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel, 2010,23(4): 289–297.
[44] Merchant AM, Zhu ZP, Yuan JQ, et al. An efficient route to human bispecific IgG. Nat Biotechnol,1998, 16(7): 677–681.
[45] Gunasekaran K, Pentony M, Shen M, et al.Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem, 2010, 285(25): 19637–19646.
[46] Muda M, Gross AW, Dawson JP, et al. Therapeutic assessment of SEED: a new engineered antibody platform designed to generate mono and bispecific antibodies. Protein Eng Des Sel, 2011, 24(5):447–454.
[47] Wranik BJ, Christensen EL, Schaefer G, et al.LUZ-Y, a novel platform for the mammalian cell production of full-length IgG-bispecific antibodies.J Biol Chem, 2012, 287(52): 43331–43339.
[48] Schaefer W, Regula JT, Bahner M, et al.Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA, 2011,108(27): 11187–11192.
[49] Doppalapudi VR, Huang J, Liu DG, et al. Chemical generation of bispecific antibodies. Proc Natl Acad Sci U S A, 2010, 107(52): 22611–22616.
[50] Chang CH, Rossi EA, Goldenberg DM. The dock and lock method: a novel platform technology for building multivalent, multifunctional structures of defined composition with retained bioactivity. Clin Cancer Res, 2007, 13(18 Pt 2): 5586s-5591s.
[51] Kipriyanov SM. Generation of bispecific and tandem diabodies. Methods Mol Biol, 2009, 562:177–193.
[52] Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol, 2007, 25(12):1369–1372.
[53] Niwa R, Hatanaka S, Shoji-Hosaka E, et al.Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 Is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res, 2004, 10(18 Pt 1): 6248–6255.
[54] Natsume A, In M, Takamura H, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res, 2008,68(10): 3863–3872.
[55] Richards JO, Karki S, Lazar GA, et al.Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells.Mol Cancer Ther, 2008, 7(8): 2517–2527.
[56] Robbie GJ, Criste R, Dall'acqua WF, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother, 2013, 57(12): 6147–6153.
[57] Stapleton NM, Andersen JT, Stemerding AM, et al.Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun, 2011, 2: 599.
[58] Labrijn AF, Buijsse AO, van den Bremer ET, et al.Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo.Nat Biotechnol, 2009, 27(8): 767–771.
[59] An ZQ, Forrest G, Moore R, et al. IgG2m4, an engineered antibody isotype with reduced Fc function. mAbs, 2009, 1(6): 572–579.
[60] Panowksi S, Bhakta S, Raab H, et al. Site-specific antibody drug conjugates for cancer therapy. mAbs,2014, 6(1): 34–45.
[61] Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol,2008, 26(8): 925–932.
[62] Shen BQ, Xu KY, Liu LN, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol, 2012, 30(2): 184–189.
[63] Hofer T, Skeffington LR, Chapman CM, et al.Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry, 2009,48(50): 12047–12057.
[64] Axup JY, Bajjuri KM, Ritland M, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci USA,2012, 109(40): 16101–16106.
[65] Boeggeman E, Ramakrishnan B, Pasek M, et al.Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: application for cell surface antigen detection. Bioconjug Chem,2009, 20(6): 1228–1236.
[66] Strop P, Liu SH, Dorywalska M, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates.Chem Biol, 2013, 20(2): 161–167.
[67] Hansel TT, Kropshofer H, Singer T, et al. The safety and side effects of monoclonal antibodies.Nat Rev Drug Discov, 2010, 9(4): 325–338.
[68] Hünig T. The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat Rev Immunol, 2012, 12(5): 317–318.
[69] Tabares P, Berr S, Romer PS, et al. Human regulatory T cells are selectively activated by low-dose application of the CD28 superagonist TGN1412/TAB08. Eur J Immunol, 2014, 44(4):1225–1236.
[70] Teachey DT, Rheingold SR, Maude SL, et al.Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood, 2013, 121(26): 5154–5157.
[71] Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol, 2012,30(21): 2691–2697.
[72] Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA, 2003, 100(14): 8372–8377.
[73] Giles FJ, Kantarjian HM, Kornblau SM, et al.MylotargTM(gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer, 2001, 92(2): 406–413.
[74] Gandhi MD, Evens AM, Fenske TS, et al.Pancreatitis in patients treated with brentuximab vedotin: a previously unrecognized serious adverse event. Blood, 2014, 123(18): 2895–2897.
[75] Yang H, Wang JC, Du JM, et al. Structural basis of immunosuppression by the therapeutic antibody daclizumab. Cell Res, 2010, 20(12): 1361–1371.
[76] Du JM, Wang H, Zhong C, et al. Structural basis for recognition of CD20 by therapeutic antibody Rituximab. J Biol Chem, 2007, 282(20):15073–15080.
[77] Li S, Wang H, Peng BZ, et al. Efalizumab binding to the LFA-1 αLI domain blocks ICAM-1 binding via steric hindrance. Proc Natl Acad Sci USA,2009, 106(11): 4349–4354.
[78] Liang SY, Dai JX, Hou S, et al. Structural basis for treating tumor necrosis factor α (TNFα)-associated diseases with the therapeutic antibody infliximab. J Biol Chem, 2013, 288(19): 13799–13807.
[79] Hu S, Liang SY, Guo HZ, et al. Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor α-associated diseases from a molecular view. J Biol Chem, 2013, 288(38): 27059–27067.
[80] Li BH, Zhao L, Guo HZ, et al. Characterization of a rituximab variant with potent antitumor activity against rituximab-resistant B-cell lymphoma.Blood, 2009, 114(24): 5007–5015.
[81] Qin WS, Feng JN, Li Y, et al. A novel domain antibody rationally designed against TNF-α using variable region of human heavy chain antibody as scaffolds to display antagonistic peptides. Mol Immunol, 2007, 44(9): 2355–2361.
[82] Xie ZG, Guo N, Yu M, et al. A new format of bispecific antibody: highly efficient heterodimerization, expression and tumor cell lysis.J Immunol Methods, 2005, 296(1/2): 95–101.
[83] Li BH, Meng YC, Zheng L, et al. Bispecific antibody to ErbB2 overcomes trastuzumab resistance through comprehensive blockade of ErbB2 heterodimerization. Cancer Res, 2013,73(21): 6471–6483.
[84] Kou G, Shi JP, Chen L, et al. A bispecific antibody effectively inhibits tumor growth and metastasis by simultaneous blocking vascular endothelial growth factor A and osteopontin. Cancer Lett, 2010,299(2): 130–136.
[85] Zhang DP, Li BH, Shi JP, et al. Suppression of tumor growth and metastasis by simultaneously blocking vascular endothelial growth factor(VEGF)-A and VEGF-C with a receptor-immunoglobulin fusion protein. Cancer Res, 2010, 70(6): 2495–2503.
[86] Li BH, Shi S, Qian WZ, et al. Development of novel tetravalent anti-CD20 antibodies with potent antitumor activity. Cancer Res, 2008, 68(7):2400–2408.
[87] Pan LQ, Wang HB, Xie ZM, et al. Novel conjugation of tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) with monomethyl auristatin E for efficient antitumor drug delivery. Adv Mater, 2013, 25(34): 4718–4722.
[88] Tan QQ, Guo QC, Fang C, et al. Characterization and comparison of commercially available TNF receptor 2-Fc fusion protein products. mAbs, 2012,4(6): 761–774.
[89] Wang Q, Li T, Wu ZG, et al. Novel VEGF decoy receptor fusion protein conbercept targeting multiple VEGF isoforms provide remarkable anti-angiogenesis effect in vivo. PLoS One, 2013,8(8): e70544.
[90] Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer, 2012, 12(4): 237–251.[91] Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol, 2013, 14(10):1014–1022.
猜你喜欢
实用肿瘤学杂志(2022年3期)2022-11-30
皮肤性病诊疗学杂志(2020年4期)2020-09-02
心肺血管病杂志(2019年1期)2019-04-22
当代化工研究(2016年9期)2016-03-20
中国卫生标准管理(2015年16期)2016-01-20
中国塑料(2015年8期)2015-10-14
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年6期)2015-07-01
癌变·畸变·突变(2015年3期)2015-02-27
郑州大学学报(理学版)(2014年2期)2014-03-01
