
氨基糖苷抗生素庆大霉素:基础研究的新进展及应用研究的新潜力
2015-03-08 06:28简心韵邓子新孙宇辉
生物工程学报 2015年6期
简心韵,邓子新,孙宇辉
庆大霉素 (Gentamicin) 是一种在临床上发挥着重要作用的氨基糖苷类抗生素(Aminoglycosides),最初由美国 Schering公司Weinstein等于 1963年从绛红小单孢菌Micromonospora purpurea以及棘孢小单孢菌Micromonospora echinospora中分离获得[1],并于1969年在美国投入使用。随后,日本协和发酵工业株式会社 (Kyowa Hakko Kogyo Co Ltd)Okachi等于 1974年从 Micromonospora sagamienisis以及来自俄罗斯的 Abrasimovskii等于 1990年从 Micromonospora purpurea var.violaceae中均分离获得庆大霉素[2–3]。1966年,我国著名的微生物学家王岳分离出庆大霉素小单孢产生菌,并且在 1969年成功工业化生产。目前所使用的庆大霉素主要成分是庆大霉素 C组分,包括庆大霉素C1、C2、C2a、C1a以C2b[4]。其结构主要是由 2-脱氧链霉胺(2-Deoxystreptamine) 在C-4与C-6上通过糖苷键与加拉糖胺 (Garosamine) 和绛红糖胺(Purpurosamine) 连接而成 (图 1)。由于具有广谱抗菌性以及速效杀菌性,并且价格低廉,因此被广泛应用于临床,一度成为治疗革兰氏阴性菌感染的首选药物。该抗生素通过与靶细胞内核糖体30S亚基16S rRNA上的氨酰基位点结合,引起mRNA发生错译,从而干扰蛋白质的合成杀死病原菌[5]。随着在临床上长期而大量的使用,庆大霉素不可避免地出现了严重的耐药性问题,同时氨基糖苷类抗生素普遍存在的耳毒性和肾毒性等副作用也限制了庆大霉素的使用[6]。虽然随着β-内酰胺类、奎洛酮类、头孢类等同样具有广谱抗菌性但副作用较少的抗生素的出现,使得庆大霉素等氨基糖苷类药物在临床上应用逐渐式微。但是对于具有多重耐药性的病原菌感染,庆大霉素仍然具有良好的治疗效果,特别是与 β-内酰胺类抗生素具有良好的协同效应使其仍然是临床上抗击病原菌严重感染的利器[7]。
因此,围绕庆大霉素所展开的相关研究一直被国内外众多学者所关注,特别是随着近年来现代生物技术水平的发展,人们对庆大霉素的认识更加深入。本文就近年来对庆大霉素的作用和耐药机制、生物合成途径和结构改造,及其新活性探索方面进行综述,并对庆大霉素的应用与发展前景进行展望。

图1 庆大霉素C组分化学结构Fig. 1 Structure of gentamicin C complex.
1 分子水平作用机制和耐药机制的新发现
早在20世纪 80年代,人们就开展了对于包括庆大霉素在内的氨基糖苷类药物作用机制的研究,加州大学圣克鲁斯分校 (University of California at Santa Cruz) 的Moazed等发现这一类抗生素的作用靶点位于细菌核糖体 30S亚基的16S rRNA[8]。近年来,随着X-Ray技术和生物大分子NMR技术的发展,使细菌核糖体及核糖体 RNA-氨基糖苷类抗生素复合物的高分辨率晶体结构得以阐明,从而为人们从分子水平上真正了解氨基糖苷类药物是如何作用于细菌核糖体及细菌的耐药性机制提供了可能,进而也为基于靶标结构设计新型氨基糖苷类药物展示了光明的前景。
1.1 作用机制的研究进展
现代生物化学与分子生物学的研究表明30S核糖体亚基与tRNA的结合是蛋白质合成的关键步骤之一。至今,已有至少两种细菌(Thermus thermophiles和Escherichia coli) 的核糖体30S亚基的晶体结构被成功报道[9-12],从其晶体结构中能清楚地辨析出与 tRNA结合的 3个位点:A (Aminoacyl)、P (Peptide)、E (Exit) 位点[13-14](图2A)。其中解码区A 位点是由3个腺嘌呤A1408、A1492、A1493在Helix44处通过2个 G-C碱基对组成的一个不对称内环[15](图2B)。在翻译过程中,核糖体在与 mRNA及其匹配的tRNA结合后,其构象会发生特定的变化[16]:1) 当mRNA未与tRNA 结合时,A位点处于静止空闲状态 (“OFF”),此时 A1492 和 A1493 隐藏于Helix44的内部;2) 当mRNA与互补配对的tRNA结合时,A位点构象则翻转为解码状态(“ON”),此时A1492和A1493翻出内环与mRNA及tRNA相互结合 (图2C),完成解码过程后再恢复至“OFF”状态[17]。正是这种被严格控制的有序的构象变化使核糖体在解码过程中精确识别并结合与 mRNA互补配对的 tRNA,从而保持蛋白质翻译的准确性[15]。
庆大霉素等氨基糖苷类药物正是通过与细菌核糖体30S亚基的16S rRNA解码区A位点特异性结合来发挥作用:2-DOS (环II) 能与A位点保守的G1494和U1495发生强烈的氢键作用;其环I能插入16S rRNA 的Helix44内部,主要与A1408、A1492和A1493形成氢键。这些特异性的相互作用都能帮助 16S rRNA形成稳定内环结构,这与“ON”状态的 A1492和A1493 翻出内环结构类似 (图 2C),长期处于“ON”状态使得非互补配对的 tRNA也能够通过A 位点,最终导致错误蛋白质的形成[18-19]。还有研究表明,除了以上这些保守的相互作用以外,不同的氨基糖类化合物由于其结构的不同对于A位点的亲和力不同,在影响核糖体构象翻转的作用上也有着不尽相同的方式,这也造成了它们活性上的差异[20]。然而,美国新泽西医学和牙科大学 (University of Medicine and Dentistry of New Jersey) 的Phlich研究组研究表明,氨基糖苷类药物与 A 位点的亲和力对杀菌能力的影响是次要的,而这种结合效应导致的A1492移动性的减弱却是决定其药效更为重要的因素[21]。除此之外,美国劳伦斯伯克利国家实验室 (Lawrence Berkeley National Laboratory)的Cate研究组和美国威尔康乃尔医学院 (Weill Cornell Medical College) 的Blanchard研究组的研究都揭示了由于氨基糖苷类抗生素与核糖体30S亚基的结合造成的核糖体构象变化对于细菌核糖体亚基迁移性会造成影响,这种作用抑制了核糖体再循环因子的结合,减慢了核糖体再循环过程从而影响蛋白质的合成过程[22-23]。

图2 细菌30S亚基16S rRNA氨基糖苷类抗生素结合位点 (A: 细菌核糖体结构;B: 细菌核糖体16S rRNA的A位点二级结构;C: 细菌30S亚基16S rRNA氨基糖苷类抗生素结合位点)[20]Fig. 2 Aminoglycosides bind in the 30S A site in Helix 44 (H44) of 16S rRNA. (A) Structure of bacterial ribosome.(B) Secondary structure of the bacterial 16S rRNA A site. (C) Aminoglycosides bind in the 30S A site of the 16S rRNA[20].
1.2 耐药机制的研究进展
目前,人们关于氨基糖苷类抗生素的耐药性机制的认识主要包括: 1) 减少对氨基糖苷类药物的吸收以及降低其在细胞内的积累; 2) 通过氨基糖苷类抗生素钝化酶 (Aminoglycosidemodifying enzymes, AME) 对氨基糖苷类抗生素进行修饰;3) 通过突变或甲基化修饰改变氨基糖苷类抗生素在细菌核糖体 30S亚基中 16S rRNA上的结合作用位点。其中后两种机制可造成更高水平的耐药性,因此,也更加引发人们的关注。
氨基糖苷类抗生素以氨基和羟基作为氢键的供体与细菌的核糖体发生一系列的相互作用,而它们同时也是病原菌中AME的作用靶点,被修饰后的氨基糖苷类抗生素对于细菌核糖体的亲和力减弱,从而产生耐药性。现已发现的钝化酶分为以下 3种:N-乙酰转移酶(N-Acetyltransferases, AACs)、O-磷酸转移酶(O-Phosphotransferases, APHs)和O-核苷转移酶(O-Adenyltransferases, ANTs),它们根据其作用位点的不同又分为了多个亚型,其分类、作用位点及在细菌中的分布见表1和图3[24]。
1987年加州大学圣克鲁斯分校的Noller研究组报道了16S rRNA上A位点的突变及特异性甲基化修饰造成细菌对氨基糖苷类抗生素高水平耐药的研究结果[25]。他们发现细菌16S rRNA的A位点A1408G的突变,造成了新霉素及庆大霉素等2-DOS类氨基糖苷类抗生素对于细菌核糖体亲和力的急剧下降甚至无法结合。而A位点上的特异性甲基化修饰是由细菌中16S rRNA甲基转移酶通过将A位点上的核苷酸甲基化修饰为m7G1405或m1A1408而发挥作用的,它们被归类为Rma超家族,且根据其修饰位点的不同又分为G1405和A1408修饰酶两类[26]。由于16S rRNA在物种中具有高度保守的特性,因此这种耐药机制在临床上可以被监测。但近年来人们在多种病原菌中发现了由质粒介导的 16S rRNA甲基转移酶,如从Escherichia coli 临床分离菌中发现的NpmA[27]和 ArmA[28],从 Poteus mirabilis分离得到的RmtC[29]等,它们均能特异性甲基化A1408,导致病原菌对氨基糖苷类抗生素多种类及高水平的耐药。这一发现也暗示了由于这种质粒介导的甲基转移酶在各个病原菌中的传播可能是造成近年来氨基糖苷类抗生素出现大面积耐药现象的原因之一。最新研究也揭示了NpmA与16S rRNA复合物的晶体结构[30],这为开发这一类甲基转移酶的抑制剂从而遏制氨基糖苷类抗生素耐药现象提供了线索。

图 3 氨基糖苷类抗生素钝化酶与卡那霉素 B的相互作用位点Fig. 3 AME target sites on kanamycin B.
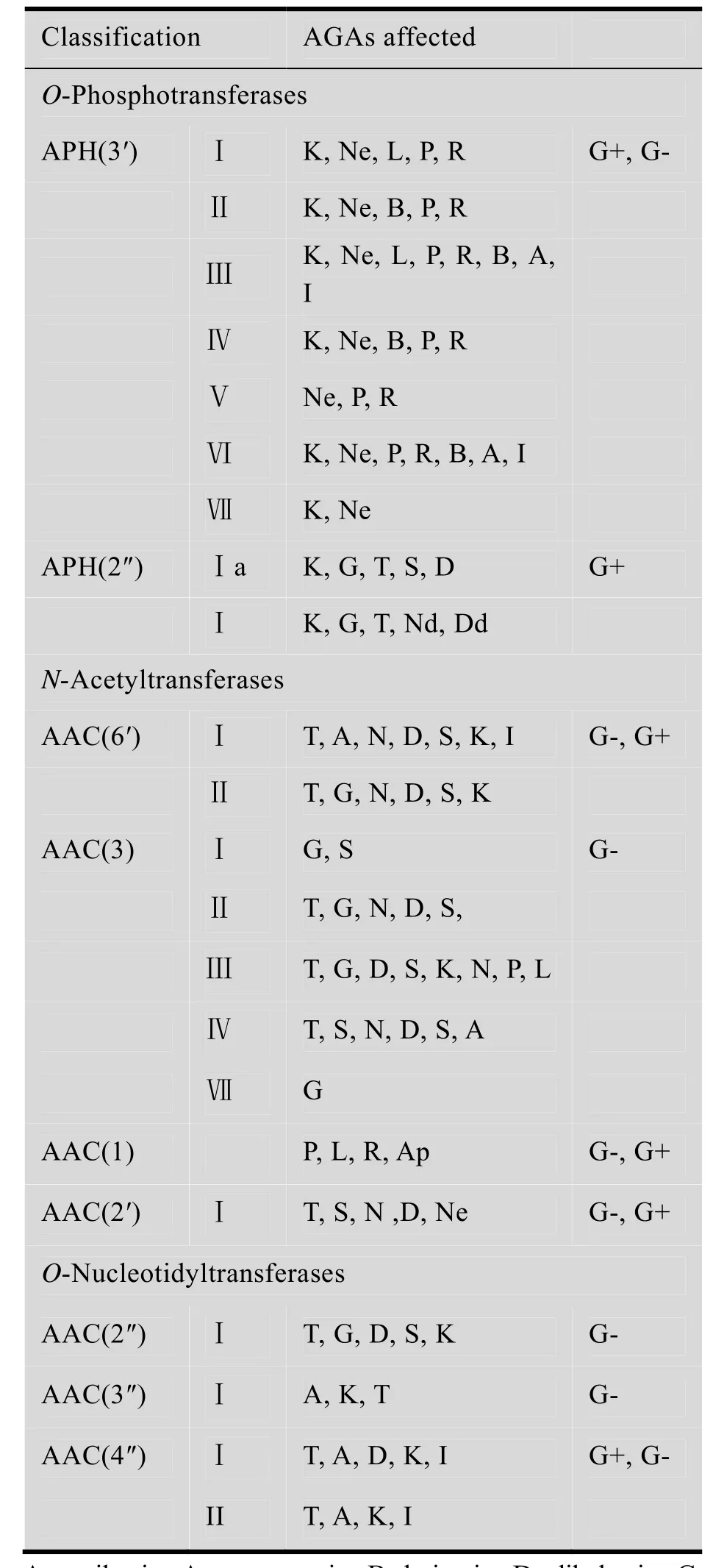
表1 主要的氨基糖苷类抗生素钝化酶分类与分布[31]Table 1 Classification and dissemination of main AME[31]
氨基糖苷类抗生素作用机制及耐药性机制在分子水平上的研究成果为基于核糖体结构设计和开发抗感染力更强、与病原RNA结合更特异、封闭钝化酶作用位点的新型氨基糖苷类抗生素衍生物及钝化酶与 16S rRNA甲基转移酶抑制剂打下了良好的基础。
2 生物合成途径的解析
一直以来人们期待能够全面清晰地阐明庆大霉素的生物合成机制,然而到目前为止,庆大霉素的生物合成途径仍然存在许多未解之谜。近年来,随着测序技术以及分子生物学技术的发展,越来越多的基因功能得以阐明,对其生物合成途径的分子水平解析也取得了长足的进步。
2.1 基于传统随机诱变突变株建立生物合成模型
早在庆大霉素发现之初,Weinstein等发现在庆大霉素产生菌的发酵产物中除了含有庆大霉素C1、C1a、C2和C2a等主要成分外,还含有一些微量但同样具有抗菌活性的组分[32-33]。随着产物分离技术以及分析检测技术水平的提高,庆大霉素 C2b以及合成中间体 A2、X2、G418、JI-20A以及JI-20B等一系列化合物相继被分离并结构鉴定[34-38]。这些组分的发现为庆大霉素生物合成精细途径的揭示奠定了基础。1967年,美国Schering公司的Testa等通过对利用传统理化方法随机诱变所获得的庆大霉素生物合成阻断突变株进行中间体喂养和生物转化试验,首次提出庆大霉素 C组分化合物通过两条分支途径进行生物合成的模型[39]。随后该研究组进一步提出可能参与庆大霉素生物合成的甲基转移、氨基转移、脱氢反应、糖基转移等步骤[40]。不过在此之后,由于受限于其产生菌分子遗传操作技术手段的建立,在较长的一段时间内,庆大霉素生物合成途径的相关研究都未能取得突破性的进展。
2.2 基于现代分子遗传学与生物化学解析生物合成途径
随着现代分子生物学与DNA测序技术的发展,从分子水平上对庆大霉素等氨基糖苷类抗生素生物合成途径的揭示逐步进入一个新的阶段。1990年,英国莱斯特大学 (University of Leicester) 的Kelemen等首次从庆大霉素产生菌Micromonospora purpurea中克隆到庆大霉素抗性基因 (grmA,即gtmF或gmrA)[41]。随后,英国华威大学 (University of Warwick) 的Wellington EM 研究组,韩国鲜文大学 (Sun Moon University) 的Sohng JK研究组以及德国伍珀塔尔大学 (Bergische Universität Wuppertal)的Piepersberg W研究组分别利用2-脱氧链霉胺生物合成基因中的保守序列作为探针,克隆到庆大霉素生物合成基因簇并完成了基因簇测序(GenBank Accession No. 分别为:AY524043、AJ575934、AJ628149)[42-44](图4),他们分别对庆大霉素生物合成基因簇相关基因进行了命名。2004年,韩国鲜文大学Kharel等通过蛋白体外催化实验,揭示出gtmA (即gntB或genC) 是负责庆大霉素生物合成的第一步反应,即葡萄糖-6-磷酸 (Glucose-6-phosphate) 转化为2-脱氧肌醇 (2-Deoxy-scyllo-inosose) 步骤的关键基因[43]。第一次将体外生化酶学方法应用于庆大霉素生物合成机制的解析。2008年,韩国梨花女子大学 (Ewha Womans University) 的Park等将基因簇中原本分散排列的参与葡萄糖-6-磷酸到庆大霉素A2生物合成的9个相关基因通过基因重组,克隆至红霉素组成型启动子 PermE*下,并在Streptomyces venezuelae YJ003中实现了异源表达,检测到目标产物庆大霉素A2。首次从分子遗传学角度证实了庆大霉素从起始合成原料葡萄糖-6-磷酸到首个类三糖中间体庆大霉素 A2的生物合成路径[45]。

图4 庆大霉素生物合成基因簇Fig. 4 Gentamicin biosynthetic gene cluster.
由于长期以来庆大霉素产生菌的分子遗传操作系统都难以建立,因此,庆大霉素生物合成机制的体内研究也一直都未有进展。直到2004年,Wellington EM研究组才首次通过基于同源重组的基因插入失活方法证实了所克隆庆大霉素基因簇[46]。随后,韩国明知大学 (Myongji University) 的Kwon HJ研究组在2008年通过基因置换的方法,将硫链丝菌素抗性基因替换庆大霉素生物合成基因簇中的甲基转移酶同源基因gntE (即gtmI或genD1) 导致突变株中间产物庆大霉素A2的积累,提出gntE是负责庆大霉素生物合成中类三糖 (Pseudotrisaccharide) 中间产物庆大霉素A2的第一步修饰的基因[46]。随后福州大学 Hong研究组同样利用基因置换的方法,将红霉素抗性基因替换庆大霉素生物合成基因簇中另一甲基转移酶同源基因 gntK (即gacD或 genK),导致突变株发酵产物不再产生在 C-6′发生甲基化修饰的庆大霉素 C组分 (C1和C2),同时明显提高了在C-6′未发生甲基化修饰的庆大霉素C组分 (C1a) 的产量,因而揭示gntK是负责C-6′甲基化修饰的基因[47]。2013年,沈阳药科大学 Xia研究组通过体内同框敲除gacD (即gntK或genK) 获得了庆大霉素C1a的高产突变株[48]。同年,美国德州大学奥斯汀分校 (University of Texas at Austin) 的Liu HW研究组在体外成功表达出钴胺素依赖型甲基转移酶GenK,并利用庆大霉素X2作为底物进行蛋白体外催化实验,成功观察到 C-6′上带有甲基化的产物G418的产生,从体外生化酶学角度证明genK是负责编码催化庆大霉素C-6′位甲基转移的基因[49]。同样通过蛋白体外催化的方法,上海医药工业研究院陈代杰研究组和中国科学院上海有机化学研究所刘文研究组利用体外表达的 GntI (即 GtmJ或 GenP) 与卡那霉素 B(Kanamycin B,庆大霉素JI-20A类似物) 进行催化反应,成功检测到在 C-3′含有磷酸化修饰的卡那霉素 B,推测 gntI也负责催化庆大霉素C-3′位磷酸转移,成为JI-20A和JI-20B脱去羟基的前提[50]。至此,人们对庆大霉素生物合成途径的研究逐渐深入至对其生物合成基因簇内各基因功能的揭示,为从分子水平全面阐释庆大霉素的合成机制开辟了道路。
2014年,武汉大学孙宇辉研究组与剑桥大学Peter Leadlay研究组综合利用分子遗传学与生物化学等方法,系统地解析了庆大霉素合成途径中从首个类三糖化合物庆大霉素 A2到庆大霉素多组分合成关键中枢分支点庆大霉素X2的生物转化过程:采用同框敲除的手段对该途径中所涉及的4个关键基因genD2 (即gntC或gtmC)、genS2 (即 gntF 或 gtmD)、genN 和 genD1分别进行基因失活,并对突变株进行中间产物喂养,根据突变株发酵产物变化预测出所敲除的目的基因与催化步骤的对应关系——genD2负责编码催化加拉糖胺 C-3″羟基氧化的脱氢酶,genS2负责编码加拉糖胺 C-3″氨基形成的氨基转移酶,genN负责编码催化加拉糖胺C-3″上氨基甲基化的甲基转移酶,genD1负责编码催化加拉糖胺 C-4″甲基化的甲基转移酶;同时证实genK编码负责催化庆大霉素A2形成庆大霉素A2e的甲基转移酶,并在ΔgenD1中发现庆大霉素Ae的积累;从 ΔgenS2ΔgenK突变株中分离出庆大霉素A2,利用其作为底物,与通过体外表达的GenD2、GenS2、GenN进行酶学催化反应可以获得庆大霉素A,然后再经过GenD1催化可以获得庆大霉素X2,从而在体外重建该生物合成途径[51]。同时,本研究组还揭示了位于分支点的庆大霉素 X2到下游不同支路的产物JI-20A和JI-20B的生物转化途径:同样采用同框敲除的方法对genQ以及genK分别进行单基因和双基因失活,同时结合对突变株进行中间产物喂养实验,证明genQ是负责编码导向不包含 C-6′甲基化修饰的庆大霉素 C组分化合物支路的基因,并验证了genK是负责编码导向含有C-6′甲基化修饰的庆大霉素 C组分化合物支路的基因;同时对 4个编码磷酸吡哆醛依赖型氨基转移酶的基因genB1 (即gntW或gacK)、genB2(即 gntL或gacE)、genB3 (即 gntJ或 gacC)、genB4(即 gntH或 gacB) 分别进行同框单基因失活以及多重基因失活,并在体外表达出这 4个基因所编码的蛋白,通过与GenQ联合催化G418反应,发现这 4个基因所编码的蛋白均可以不同程度的催化绛红糖胺的 C-6′的氨基转移过程,其中GenB1催化效率最高;另外还发现GenB2具有催化庆大霉素 C2a与 C2异构化作用,GenB3与GenB4很可能参与绛红糖胺的C-3′与C-4′上脱羟基过程[52]。上述研究以庆大霉素生物合成途径中关键分支点庆大霉素X2为突破口,全面地揭示出了庆大霉素X2的合成来源以及通往下游产物的去路 (图5),为利用组合生物合成的方法和技术开展庆大霉素高效定向优化和创 新提供了理论依据。同年,沈阳药科大学夏焕章研究组通过体内同框敲除gacJ (即gntX或genQ)并辅以随机诱变的方式获得G418高产突变株[53],为利用庆大霉素产生菌生物发酵直接获得单一组分产物探索了新的方法。
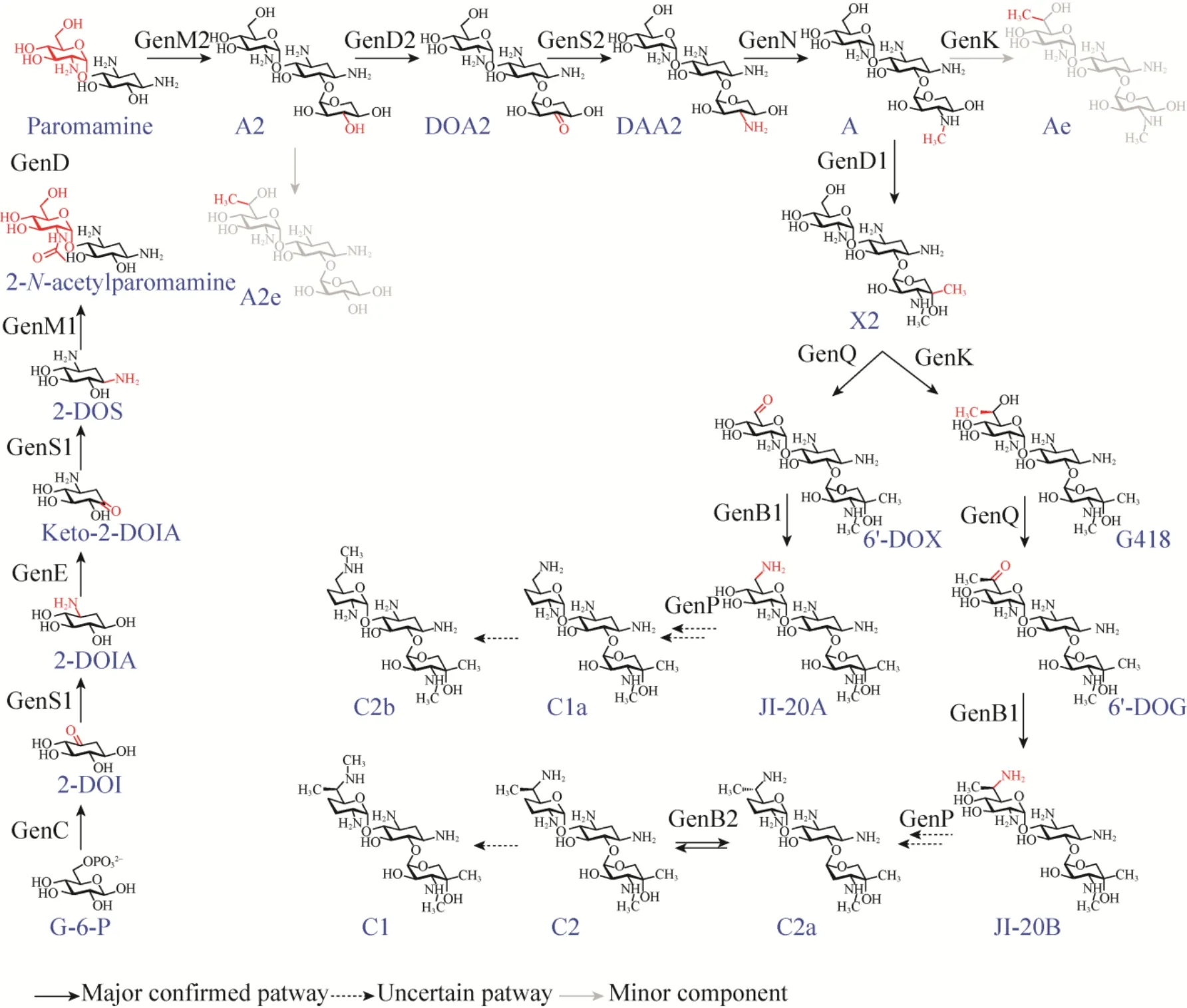
图5 目前已知的庆大霉素生物合成途径Fig. 5 Currently confirmed gentamicin biosynthetic pathway.
3 新型氨基糖苷类抗生素的研发及新生物活性的揭示
3.1 基于核糖体结构及AME作用机制设计的半合成新型氨基糖苷类抗生素的研发
以庆大霉素为代表的第一代氨基糖苷类抗生素曾被广泛应用于临床上,但随之而来的耐药性及毒性问题也引起了人们的密切关注。随着对氨基糖苷类抗生素作用机制与耐药性机制研究的深入,结合核糖体结构与AME作用机制研究而设计的第二代半合成氨基糖苷类抗生素衍生物的开发也随之进入了高潮,如:基于卡那霉素改造而来的地贝卡星 (Dibekacin,1971年)、阿米卡星 (Amikacin,1972年)、阿贝卡星(Arbekacin,1973年) 。基于庆大霉素和西索米星(Sisomicin) 改造而来的异帕米星(Isepamicin,1975年)、奈替米星 (Netilmicin,1976年) (图6) 都已成功投放于市场。
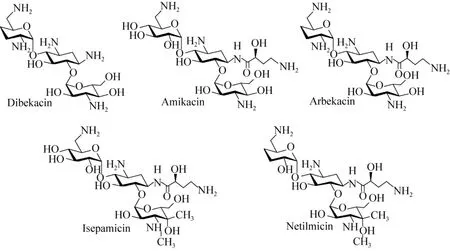
图6 第二代半合成氨基糖苷类抗生素衍生物结构Fig. 6 Structure of the second generation of aminoglycosides.

图7 Plazomicin结构及其封闭的氨基糖苷类抗生素钝化酶作用位点[54]Fig. 7 Structure of Plazomicin in relation to AME target sites[54].
虽然在上世纪70年代末,由于喹诺酮类抗生素广泛应用使得氨基糖苷类抗生素的使用大幅度地减少,但近年来作为能抗击由多重耐药菌引起的严重感染的重要药物,氨基糖苷类药物又重新受到了医学界的重视。目前,人们将研究重点集中在能阻断 AME作用位点及低毒性的第三代氨基糖苷类抗生素开发上。由美国Achaogen公司开发的Plazomicin (ACHN-490)[54](图 7) 则是目前最为典型的代表,它以西索米星为前体通过化学合成而来,结合了多种氨基糖苷类抗生素的结构特点以增强其抗菌活性,封闭了重要的 AME作用位点与毒性位点:在2-DOS上引入来自于新霉素的 4-氨基-2-羟基丁酰 (4-Amino-2-hydroxybutyric acid,AHBA)侧链显著增强了其抗菌活性;环III上引入来自于庆大霉素的甲基化修饰,使其躲避了钝化酶AAC的修饰;环I上3,4位同于西索米星双羟基的缺失的结构特点也能抵御来自于钝化酶APH (3′)、ANT (4′)的修饰;环 I 6′-NH3 上引入的羟乙基也封闭了AAC (6′)的作用位点及氨基糖苷类抗生素毒性来源的主要位点之一。该抗生素以其能抗击多重耐药菌及低毒的特性已于2012年成功完成了第二阶段的临床试验[55]。
3.2 新生物活性与新药用潜力的挖掘
伴随着对氨基糖苷类药物作用机制的深入研究,其新的生物活性也不断被揭示。法国国家科学研究院分子和细胞生物学研究所(Institut de Biologie Moléculaire et Cellulaire du CNRS) 的Westhof E研究组发现庆大霉素等氨基糖苷类药物能够与核糖体内RNA特定部位通过不规则碱基配对形成的“内环”结构相结合,而这种特殊的内环结构与多种病毒RNA的复制密切相关,同时它们还存在于HIV的效应元件结合区中[56],这一现象引发了人们对于开发特异性抗病毒的氨基糖苷类抗生素的极大兴趣。目前研究较多的能与氨基糖苷类药物结合的特异性靶标RNA及RNA调节因子有:核酶Ⅰ型内含子、锤头核酶和丁型肝炎核酶;HIV反式激活效应元件 (Trans-activating responds element,TAR),rev效应元件 (Rev-response element,RRE)。这些研究在治疗与RNA相关的疾病方面,具有潜在的应用价值,使得氨基糖苷类药物及其衍生物有望用于艾滋病、肝炎病毒等疾病的治疗[57]。
碱基对的置换、插入或者缺失产生的非成熟终止密码子 (Premature termination codon,PTC) 可造成肽链的合成提前终止,形成没有功能或者是功能不完整的蛋白质,这是导致 12%的人类遗传疾病发生的根源[58]。1996年,美国阿拉巴马大学伯明翰分校 (University of Alabama at Birmingham) Howard等在对囊性纤维化 (Cystic fibrosis,CF) 的研究过程中首次观察到庆大霉素等氨基糖苷类化合物可以在哺乳动物细胞内抑制 PTC,从而合成完整的有功的蛋白质,该研究结果暗示着庆大霉素可以用于治疗囊性纤维化[59]。随后,具有AHBA侧链的巴龙霉素 (Butirosin) 衍生物 NB30、NB54及NB84 也相继被开发出来,体外细胞培养观察到这3种衍生物对CF、进行性假肥大性肌营养不良 (Duchenne muscular dystrophy,DMD) 及Rett综合征的PTC均有抑制作用[60-62]。2010 年,美国俄亥俄州立大学医学院 (The Ohio State University College of Medicine) 的Mendell JR研究组发现庆大霉素能改善由无义突变引起的DMD症状[63];此外,研究表明庆大霉素在治疗遗传性心率失常方面,也具有一定治疗活性[64]。
p53是一类重要的肿瘤抑制因子,其突变与癌症的发生有着密切的关系。近期也有研究报道显示,在含有 p53无义突变的人乳腺癌培养细胞中加入不同的氨基糖苷类抗生素 (G418、庆大霉素等),能够恢复PTC引起的肿瘤抑制因子 p53蛋白质的不完全翻译并且使肿瘤细胞的凋亡数增加。这一研究的发现为氨基糖苷类药物利用终止密码子的通过效应来抑制肿瘤细胞生长提供了理论依据[65],使得将氨基糖苷类药物应用于肿瘤的治疗有了突破性的进展。
最新研究发现,低剂量的庆大霉素还可以作为一种增敏剂,在体外能够显著提高抗肿瘤药物喜树碱、洋地黄毒苷和长春花碱对NCI-H460肺癌细胞的作用活性[66],这项研究为氨基糖类抗生素新药用功能的挖掘提供了新的思路。
因此,研制和开发新型氨基糖苷类药物或其衍生物,用于由治疗无义突变引起的人类遗传性疾病及利用终止密码子的通过效应来发挥的抗肿瘤活性成为了氨基糖类抗生素老药新用的发展方向之一。结合对氨基糖苷类药物药理学的研究,不断深入挖掘其新的药用活性也能为氨基糖苷类药物重新注入新的血液。
4 总结与展望
庆大霉素自进入临床使用以来,以其良好的治疗效果一直在临床治疗中发挥着重要的作用。虽然因毒性及耐药性问题受到了一定的限制,但作为治疗由革兰氏阴性菌导致的严重感染的首选药物在临床中仍然占据着不可替代的位置。随着近年来分子生物学技术的发展,使人们在分子水平对其作用机制、耐药性机制、生物合成路径有了更深入的了解,涌现出了一批新型的氨基糖苷类抗生素衍生物,并揭示出了其新的活性,在新的药用功能的挖掘中也发现了其巨大的潜力。
人们期待着庆大霉素等氨基糖苷抗生素未来将有可能从几个方面取得突破:1) 对庆大霉素生物全合成途径的最终阐明,包括对目前仍然未知的绛红糖胺 C-3′和 C-4′上脱双羟基过程以及绛红糖胺的 N-6′上甲基化过程的揭示。这些生物合成途径的揭示有助于我们更好地利用庆大霉素,对其产生菌进行基因定向改造,提高庆大霉素产量或者产生单一组分化合物。2)基于对作用机制与耐药性机制的了解,利用组合生物合成和合成生物学的手段对庆大霉素产生菌进行分子水平层面的改造,引入新的生物合成模块,使其产生非天然结构同时又具有新活性,特别是针对耐药性菌株的新氨基糖苷化合物。3) 基于特异性结合 RNA的特点定向筛选或改造,开发出具有低毒或无毒副作用的新型庆大霉素衍生物,使得传统老药焕发新的活力。
[1] Weinstein MJ, Luedemann GM, Oden EM, et al.Gentamicin, new antibiotic complex from Micromonospora. J Med Chem, 1963, 6(7):463–464.
[2] Okachi R, Kawamoto I, Takasawa S, et al. A new antibiotic XK-62-2 (Sagamicin). I. Isolation,physicochemical and antibacterial properties. J Antibiot (Tokyo), 1974, 27(10): 793–800.
[3] Abrasimovskii PI, Valdimirov AV, Bartoschevich IE. Phosphate regulation of the processes of growth and biosynthesis of gentamicin in Micromonospora purpurea var. violaceae. Antibiot Khimioter,1990, 35(1): 5–8.
[4] Wagman GH, Oden EM, Weinstein MJ. Differential chromatographic bioassay for the gentamicin complex. Appl Microbiol, 1968, 16(4): 624–627.
[5] Fourmy D, Recht MI, Blanchard SC, et al.Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science, 1996, 274(5291): 1367–1371.
[6] Mathews A, Bailie GR. Clinical pharmacokinetics toxicity and cost effectiveness analysis of aminoglycosides and aminoglycoside dosing services. J Clin Pharm Ther, 1987, 12(5): 273–291.
[7] Shaw KJ, Rather PN, Hare RS, et al. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycosidemodifying enzymes. Microbiol Rev, 1993, 57(1):138–163.
[8] Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA.Nature, 1987, 327(6121): 389–394.
[9] Wimberly BT1, Brodersen DE, Clemons WM Jr, et al. Structure of the 30S ribosomal subunit. Nature,2000, 407(6802): 327–339.
[10] Carter AP, Clemons WM, Brodersen DE, et al.Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature, 2000, 407(6802): 340–348.
[11] Ogle JM, Brodersen DE, Clemons WM Jr, et al.Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science, 2001, 292(5518):897–902.
[12] Vila-Sanjurjo A, Ridgeway WK, Seymaner V, et al.X-ray crystal structures of the WT and a hyperaccurate ribosome from Escherichia coli. Proc Natl Acad Sci USA, 2003, 100(15): 8682–8687.
[13] Green R, Noller HF. Ribosomes and translation.Annu Rev Biochem, 1997, 66: 679–716.
[14] Tsai A, Uemura S, Johansson M, et al. The impact of aminoglycosides on the dynamics of translation elongation. Cell Rep, 2013, 3(2): 497–508.
[15] Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon-anticodon helix by ribosomal RNA.Science, 1999, 285(5434): 1722–1725.
[16] Nierhaus KH. Solution of the ribosome riddle: how the ribosome selects the correct aminoacyl-tRNA out of 41 similar contestants. Mol Microbiol, 1993,9(4): 661–669.
[17] Ogle JM, Brodersen DE, Clemons WM Jr, et al.Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science, 2001, 292(5518):897–902.
[18] Shandrick S, Zhao Q, Han Q, et al. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem Int Ed Engl, 2004, 43(24):3177–3182.
[19] Kaul M, Barbieri CM, Pilch DS. Fluorescencebased approach for detecting and characterizing antibiotic-induced conformational changes in ribosomal RNA: comparing aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences. J Am Chem Soc, 2004, 126(11):3447–3453.
[20] Feldman MB, Terry DS, Altman RB, et al.Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nat Chem Biol, 2010, 6(3): 244.
[21] Kaul M, Barbieri CM, Pilch DS.Aminoglycoside-induced reduction in nucleotide mobility at the ribosomal RNA A-site as a potentially key determinant of antibacterial activity.J Am Chem Soc, 2006, 128(4): 1261–1271.
[22] Borovinskaya MA, Pai RD, Zhang W, et al.Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol,2007, 14(8): 727–732.
[23] Wang L, Pulk A, Wasserman MR, et al. Allosteric control of the ribosome by small-molecule antibiotic. Nat Struct Mol Biol, 2012, 19(9):957–963.
[24] Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug Resist Updat, 2010,13(6): 151–171.
[25] Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA.Nature, 1987, 327(6121): 389–394.
[26] Doi Y, Arakawa Y. 16S ribosomal RNA methylation: emerging resistance mechanism against aminoglycosides. Clin Infect Dis, 2007,45(1): 88–94.
[27] Wachino J, Shibayama K, Kurokawa H, et al. Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob Agents Chemother, 2007, 51(12): 4401–4409.
[28] González-Zorn B, Teshager T, Casas M, et al. armA and aminoglycoside resistance in Escherichia coli,Emerging Infect Dis, 2005, 11(6): 954–956.
[29] Wachino J, Yamane K, Shibayama K, et al. Novel plasmid-mediated 16S rRNA methylase, RmtC,found in a Proteus mirabilis isolate demonstrating extraordinary high-level resistance against various aminoglycosides. Antimicrob Agents Chemother,2006, 50(1): 178–184.
[30] Dunkle JA, Vinal K, Desai PM, et al. Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc Natl Acad Sci USA, 2014, 111(17):6275–6280.
[31] Becker B, Cooper MA. Aminoglycoside antibiotics in the 21st century. ACS Chem Biol, 2013, 8(1):105–115.
[32] Weinstein MJ, Wagman GH, Oden EM, et al.Biological activity of the antibiotic components of the gentamicin complex. J Bacteriol, 1967, 94(3):789–790.
[33] Wagman GH, Marquez JA, Weinstein MJ.Chromatographic separation of the components of the gentamicin complex. J Chromatogr, 1968,34(2): 210–215.
[34] Wagman GH, Marquez JA, Bailey JV, et al.Chromatographic separation of some minor components of the gentamicin complex. J Chromatogr, 1972, 70(1): 171–173.
[35] Daniels PJ, Luce C, Nagabhushan TL. The gentamicin antibiotics. 6. gentamicin C2b, an aminoglycoside antibiotic produced by Micromonospora purpurea mutant JI-33. J Antibiot (Tokyo), 1975, 28(1): 35–41.
[36] Bérdy J, Pauncz JK, Vajna ZM, et al. Metabolites of gentamicin-producing Micromonospora species I. isolation and identification of metabolites. J Antibiot (Tokyo), 1977, 30(11): 945–954.
[37] Wagman GH, Testa RT, Marquez JA, et al.Antibiotic G-418, a new Micromonospora-produced aminoglycoside with activity against protozoa and helminths:fermentation, isolation, and preliminary characterization. Antimicrob Agents Chemother,1974, 6(2): 144–149.
[38] lavsky J, Bayan AP, Charney W, et al. Antibiotic from Micromonospora poupurea JI-20: US,3986929. 1975-06-09.
[39] Testa RT, Tilley BC. Biotransformation, a new approach to aminoglycoside biosynthesis: Ⅱgentamicin, J Antibiot (Tokyo), 1976, 29(2):140–146.
[40] Testa RT, Wagman GH, Daniels PJ, et al.Mutamicins: biosynthetically created new sisomicin analogues. J Antibiot (Tokyo), 1974,279(12): 917–921.
[41] Kelemen GH, Cundliffe E, Financsek I. Cloning and characterization of gentamicin-resistance genes from Micromonospora purpurea and Micromonospora rosea. Gene, 1991, 98(1): 53–60.[42] Unwin J, Standage S, Alexander D, et al. Gene cluster in Micromonospora echinospora ATCC15835 for the biosynthesis of the gentamicin C complex. J Antibiot (Tokyo), 2004, 57(7):436–445.
[43] Kharel MK, Basnet DB, Lee HC, et al. Molecular cloning and characterization of a 2-deoxystreptamine biosynthetic gene cluster in gentamicin-producing Micromonospora echinospora ATCC15835. Mol Cells, 2004, 18(1):71–78.
[44] Aboshanab KMA. Genetic studies on the biosynthesis of the major aminoglycoside antibiotics: isolation, analysis and comparison of the biosynthetic gene clusters for 12 aminoglycoside antibiotics[D]. Wuppertal:Bergische Universität Wuppertal, 2005.
[45] Park JW, Joong JS, Parajuli N, et al. Genetic dissection of the biosynthetic route to gentamicn A2 by heterologous expression of its minimal gene set. Proc Natl Acad Sci USA, 2008, 105(24):8399–8404.
[46] Kim JY, Suh J W, Kang SH, et al. Gene inactivation study of gntE reveals its role in the first step of pseudotrisaccharide modification in gentamicin biosynthesis. Biochem Biophys Res Commun,2008, 372(4): 730–734.
[47] Hong WR, Yan L. Identification of gntK, a gene required for the methylation of purpurosamine C-6'in gentamicin biosynthesis. J Gen Appl Microbiol,2012, 58(5): 349–356.
[48] Li D, Li H, Ni X, et al. Construction of a gentamicin C1a-overproducing strain of Micromonospora purpurea by inactivation of the gacD gene. Microbiol Res, 2013, 168(5): 263–267.
[49] Kim HJ, McCarty RM, Ogasawara Y, et al.GenK-catalyzed C-6' methylation in the biosynthesis of gentamicin: isolation and characterization of a cobalamin-dependent radical SAM enzyme. J Am Chem Soc, 2013, 135(22):8093–8096.
[50] Shao L, Chen J, Wang C, et al. Characterization of a key aminoglycoside phosphotransferase in gentamicin biosynthesis. Bioorg Med Chem Lett,2013, 23(5): 1438–1441.
[51] Huang C, Huang F, Eileen M, et al. Delineating the biosynthesis of gentamicin X2, the common precursor of the gentamicin C antibiotic complex.Chem Biol, 2014, (In press)
[52] Guo J, Huang F, Huang C, et al. Specificity and promiscuity at the branch point in gentamicin biosynthesis. Chem Biol, 2014, 21(5): 608–618.
[53] Ni X, Sun Z, Zhang H, et al. Genetic engineering combined with random mutagenesis to enhance G418 production in Micromonospora echinospora.J Ind Microbiol Biotechnol, 2014, 41(9):1383–1390.
[54] Aggen JB, Armstrong ES, Goldblum AA, et al.Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother, 2010,54(11): 4636–4642.
[55] Zhanel GG, Lawson CD, Zelenitsky S, et al.Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther, 2012, 10(4):459–473.
[56] Hermann T, Westhof E. Saccharide-RNA recognition. Biopolymers. 1998, 48(2/3): 155–165.[57] Tor Y, Hermann T, Westhof E. Deciphering RNA recognition: aminoglycoside binding to the hammerhead ribozyme. Chem Biol, 1998, 5(11):R277–283.
[58] Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs, 2009, 23(3): 165–174.
[59] Howard M, Frizzell RA, Bedwell DM.Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med,1996, 2(4): 467–469.
[60] Nudelman I, Rebibo-Sabbah A, Cherniavsky M, et al. Development of novel aminoglycoside (NB54)with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem, 2009, 52(9): 2836–2845.
[61] Vecsler M, Ben Zeev B, Nudelman I, et al. Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations.PLoS ONE, 2011, 6(6): e20733.
[62] Brendel C, Belakhov V, Werner H, et al.Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med(Berl), 2011, 89(4): 389–398.
[63] Malik V, Rodino-Klapac LR, Viollet L, et al.Aminoglycoside-induced mutation suppression(stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther Adv Neurol Disord, 2010, 3(6): 379–389.
[64] Yao Y, Teng S, Li N, et al. Aminoglycoside antibiotics restore functional expression of truncated HERG channels produced by nonsense mutations. Heart Rhythm, 2009, 6(4): 553–560.
[65] Floquet C, Deforges J, Rousset JP, et al. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res, 2011, 39(8):3350–3362.
[66] Cuccarese MF, Singh A, Amiji M, et al. A novel use of gentamicin in the ROS-mediated sensitization of NCI-H460 lung cancer cells to various anticancer agents. ACS Chem Biol, 2013, 8(12): 2771–2777.
猜你喜欢
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
世界最新医学信息文摘(2021年12期)2021-06-09
中国饲料(2020年3期)2020-12-19
肿瘤防治研究(2020年5期)2020-07-09
生物学教学(2019年9期)2019-09-23
畜牧与饲料科学(2018年9期)2018-02-12
国外医药(抗生素分册)(2016年1期)2016-07-10
中外医疗(2015年11期)2016-01-04
